Publications
Publications at UA
|
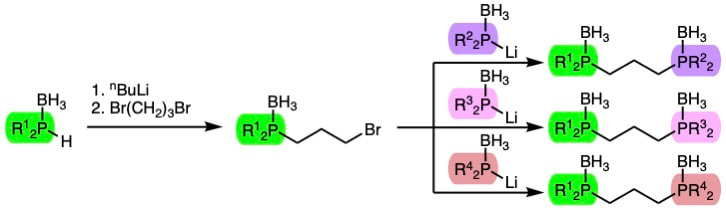
58. "Modular synthesis of unsymmetric 1,3-diphosphinopropanes through sequential substitution reactions," Headford, B. R.; Kuhnel, W. B.; Qu, F.; Shaughnessy, K. H., J. Organomet. Chem., 2023, 999, 122816.
Methods to synthesize diphosphines with different substituents on the two phosphorus centers are limited, particularly for the synthesis of unsymmetric bis(dialkylphosphine) ligands. Herein is reported a modular and high-yielding synthesis of borane-protected unsymmetric 1,3-bis(diphosphino)propanes containing either two dialkylphosphine moieties or one dialkylphosphine and one diphenylphosphine group. The method relies on sequential phosphine substitution on 1,3-dibromopropane. The borane-protected diphosphines are readily converted to bis(phosphonium salts), which serve as convenient preligands. Palladium(II) complexes of di-tert-butyl(3-(dicyclohexylphosphino)propyl)phosphine (tBuCyPP), di-tert-butyl(3-(diisobutylphosphino)propyl)phosphine (tBuiBuPP), and diisobutyl((3-(dicyclohexylphosphino)propyl)phosphine (CyiBuPP) have been prepared. The tBuiBuPP and CyiBuPP complexes have been structurally characterized and their structural properties compared to symmetric (1,3-diphosphinopropane)palladium dichloride structures. |
|
|
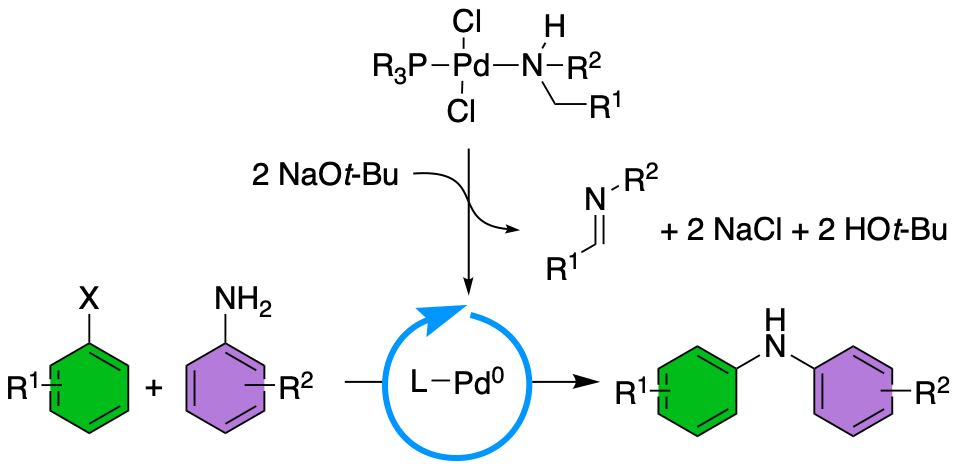
57. "Amines as activating ligands for phosphine palladium(II) precatalysts: effect of amine ligand identity on catalyst efficiency," Pierce, J. K.; Hiatt, L. D.; Howard, J. R.; Hu, H.; Qu, F.; Shaughnessy, K. H. Organometallics, 2022, 41, 3861-3871.
Palladium-catalyzed cross coupling reactions between C-X/C-H bonds and nucleophilic reagents remain a staple in organic synthesis. These reactions rely on the in situ generation of an active LPd0 species. Air-stable palladium(II) precatalysts that readily form the LPd0 species under catalytic conditions have been identified as effective precatalysts. Alkyl amine ligand can serve as potential reducing ligands for palladium(II) complexes in the presence of base through a sequence of β-hydrogen elimination and base-promoted reductive elimination of the resulting palladium hydride. A series of air-stable (tBu2PNp)Pd(amine)Cl2 (Np = neopentyl) complexes were synthesized with a range of structurally different primary and secondary amine ligands. These complexes allowed the effect of the amine ligand structure on catalyst performance in cross-coupling of aryl halides and aniline derivatives to be analyzed. The identity of the amine ligand has a significant effect on catalyst activity and lifetime. Precatalysts with primary n-alkylamine ligands afford the most active catalysts. Reactions using (tBu2PNp)Pd(n-butylamine)Cl2 as the precatalyst provide excellent yields at room temperature in under 1 hour. Moreover, (Np3P)Pd(n-butylamine)Cl2 was synthesized and exhibited promising activity for sterically demanding aniline derivatives and aryl chlorides. Preliminary mechanistic studies show the structure of the amine ligand has a significant effect on the selectivity for palladium(0) formation that correlates with the activity of the precatalysts. |
|
|
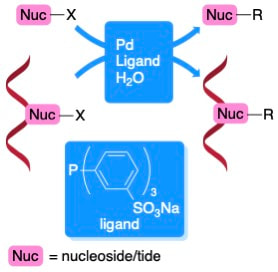
56. “Covalent Modification of Nucleobases using Water-Soluble Palladium Catalysts,” Shaughnessy, K. H. Chem. Rec. 2022, e202200190.
Nucleosides represent one of the key building blocks of biochemistry. There is significant interest in the synthesis of nucleoside-derived materials for applications as probes, biochemical models, and pharmaceuticals. Palladium-catalyzed cross-coupling reactions are effective methods for making covalent modification of carbon and nitrogen sites on nucleobases under mild conditions. Water-soluble catalysts derived from palladium and hydrophilic ligands, such as tris(3-sulfonatophenyl)phosphine trisodium (TPPTS), are efficient catalysts for a range of coupling reactions of unprotected halonucleosides. Over the past two decades, these methods have been extended to direct functionalization of halonucleotides, as well as RNA and DNA oligonucleotides (ONs) containing halogenated bases. These methods can be run under biocompatible conditions, including examples of Suzuki coupling of modified DNA in whole cells and tissue samples. In this account, development of this methodology by our group and others is highlighted along with the extension of these catalyst systems to modification of nucleotides and ONs. |
|
|
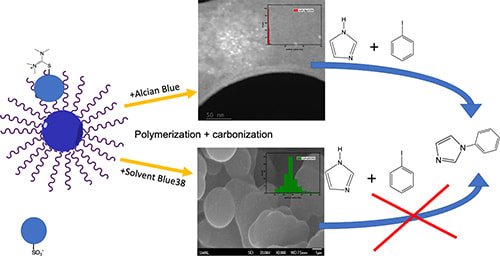
55. "Impact of copper phthalocyanine structure on catalytic activity when incorporated into hierarchically porous carbon," Adhikari, R.; Lockhart, M.; Shrestha, A.; Curley, S.; Hu, H.; Shaughnessy, K. H.; Bowman, M. K.; Bakker, M. G. Mol. Catal., 2022, 529, 11255.
A copper porphyrin and two copper phthalocyanine complexes were incorporated into a hierarchically porous carbon support formed by a sol-gel resorcinol-formaldehyde polymerization templated by F-127 block copolymer. The F127 self-organization leads to formation of mesostructure that on carbonization produces mesopores. Striking differences in catalytic activity for iodobenzene/imidazole cross coupling were observed between Alcian Blue-8gx (CuPc-8gx) (active) and copper phthalocyanine disulfonate (CuPc2S) (inactive). FTIR and EPR spectroscopy confirmed that phthalocyanine remained in the carbon after it was formed by heating to 500 °C, but that no complex survived heating to 800 °C. Electron microscopy (SEM and TEM) showed that the two dyes were differently distributed within the carbon support and found very different sizes of copper nanoparticles: 1.5 nm for CuPc-8gx and 86 nm for CuPc2S. Based on quenching of pyrene fluorescence by the two dyes it was concluded that CuPc-8gx was incorporated into F127 micelles. During polymerization the CuPc-8gx remained associated with F127 and so was incorporated into the polymer and subsequently the carbon. CuPc2S was not associated with F127 micelles, was not incorporated into the polymer and instead precipitated out during drying. This led to large CuPc2S nanoparticles, which resulted in very large copper nanoparticles with no catalytic activity. |
|
|
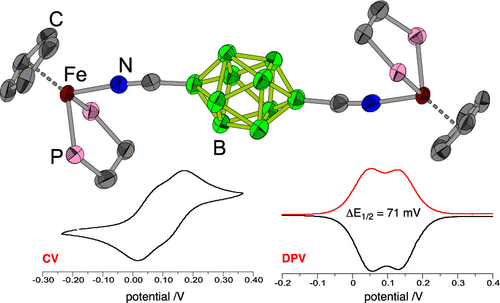
54. "Decaborate [closo-B10H10]2– as an efficient conduit of electronic effects: Comparative studies of Fe…Fe communication in [{(η5-Cp)(dppe)Fe}2{μ2-(NC-X-CN)}]n+ (n =0,2)," Guschlbauer, J.; Shaughnessy, K. H.; Pietrzak, A.; Chung, M.-C.; Sponsler, M. B.; Kaszyński, P. Organometallics, 2021, published online.
Electronic communication between Fe centers in the diiron complexes [{(η5-Cp)(dppe)Fe}2{μ2-(NC-X-CN)}]n+ (n = 0, 2) in which X = [−B10H8−]2–, −C6H4–, −C8H12–, and −C2B10H10– was investigated by spectroscopic (UV–vis and IR), electrochemical (CV and DPV), and single-crystal XRD methods. All experimental results were supported by a DFT computational analysis (B3LYP/Def2SVP) of model complexes that included two other closo-boranes: X = −C2B8H8– and [−B12H10−]2–. The DFT analysis focused on the geometry, electronic excitations, IR spectra, charge and spin delocalization, and intramolecular spin–spin exchange interactions in the series of all six complexes and their singly and doubly oxidized analogues. The results demonstrate that [closo-B10H8-1,10-diyl]2– has the best ability among the closo-boranes to mediate electronic effects similar to those of 1,4-phenylene. This is evident from the observed separation of one-electron-oxidation waves (∼70 mV) and comparable S–T energy gaps in the doubly oxidized species. |
|
|
53. "How addition of a nickel cyclohexyl-salen complex impacts a one-pot synthesis of nickel/hierarchically porous carbon monolith catalyst," Adhikari, R.; Kotbagi, T. V.; Shaughnessy, K. H.; Sherwood, J.; Bao, Y.; Bakker, Martin G.* J. Sol-Gel Sci. Technol., 2021, 106-116.
|
|
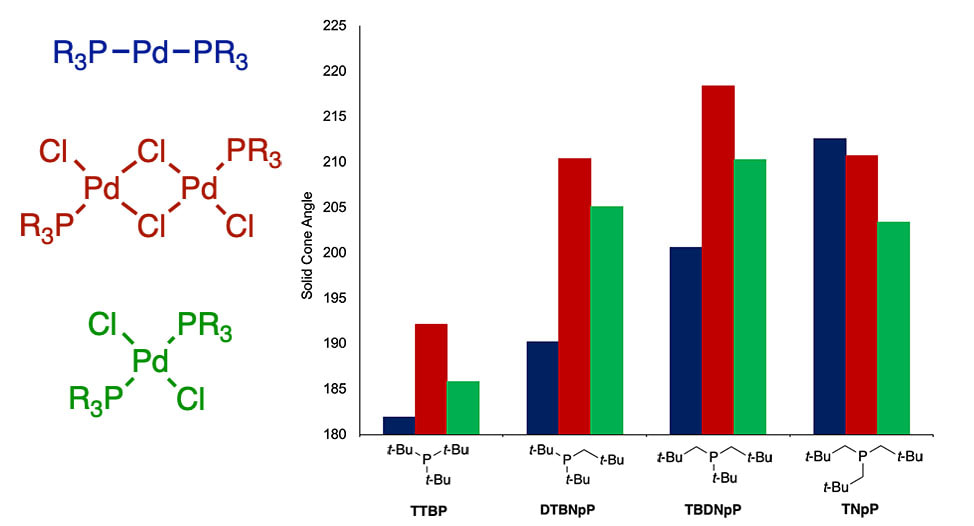
52. "Experimental and Computational Study of the Structure, Steric Properties, and Binding Equilibria of Neopentylphosphine Palladium Complexes," Barnett, K. L.; Vasiliu, M.; Stein, T. H.; Delahay, M. V.; Qu, F.; Gerlach, Deidra L.; Dixon, D. A.; Shaughnessy, K. H. Inorg. Chem. 2020, 59, 5579-5592.
Steric properties of crystallographically and computationally determined structures of linear palladium(0) and square planar palladium(II) complexes of di-(tert-butyl)neopentylphosphine (P(t-Bu)2Np) tert-butyldineopentylphosphine (P(t-Bu)Np2) and trineopentylphosphine (PNp3) have been determined. Structures of linear palladium(0) complexes show that steric demand increases as tert-butyl groups are replaced with neopentyl groups (P(t-Bu)2Np < P(t-Bu)Np2 < PNp3). In square planar palladium(II) complexes, PNp3 gives the smallest steric parameters, whereas P(t-Bu)Np2 has the largest steric demand. The change in the steric demand of PNp3 compared to P(t-Bu)2Np and P(t-Bu)Np2 results from a significant conformational change in PNp3 depending on the coordination number of the metal. The steric properties of these ligands were also probed by measuring the equilibrium constant for coordination of free phosphine to dimeric [(R3P)Pd(μ-Cl)Cl]2 complexes. Binding equilibria follow the same trend as the steric parameters for square planar complexes with PNp3 having the highest binding constant. In contrast to the normal trend, the neopentylphosphines show increased pyramidalization at phosphorus with increasing steric demand. We hypothesize that this unusual dependence reflects the low back side strain of the neopentyl group, which allows the ligand to be more pyramidalized while still exerting a significant front side steric demand. |
|
|
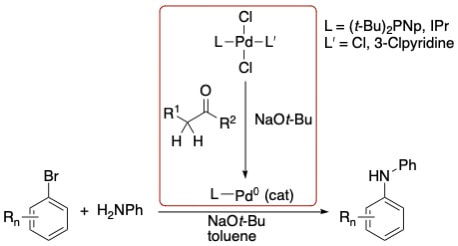
51. "Enolizable Ketones as Activators of Palladium(II) Precatalysts in Amine Arylation Reactions," Hu, H.; Gilliam, A. M.; Qu, F.; Shaughnessy, K. H. ACS Catal., 2020, 10, 4127-4135.
Enolizable ketones have been identified as effective activators for palladium(II) precatalysts in the coupling of aryl bromides and aniline. N-Arylation reactions catalyzed by [(DTBNpP)PdCl2]2 (DTBNpP = (di-(tert-butyl)neopentylphosphine) and PEPPSI-IPR precatalysts are activated by the addition of acetone, mesityl oxide, and 3-pentanone. 3-Pentanone was the most effective activator. Mechanistic studies show that acetone, 3-pentanone, and mesityl oxide reduce [(DTBNpP)PdCl2]2 in the presence of NaOt-Bu to Pd(0)(DTBNpP)2. |
|
|
50. "Development of Palladium Precatalysts that Efficiently Generate LPd(0) Active Species," Shaughnessy, K. H. Isr. J. Chem. 2020, 60, 180-194.
Invited review Included in Wiley Hot Topic: C-C Coupling. Palladium catalysts have become central to modern organic synthesis, particularly in the context of cross-coupling reactions to form C–C and C-heteroatom bonds. The development of highly efficient catalyst systems has seen a transition from the use of in situ-generated catalysts derived from a ligand source and palladium precursor to the use of preformed palladium-ligand complexes that can efficiently generate the active species. The design of these systems has focused on optimizing the generation of the LPd(0) species presumed to be the active catalysts with most state-of-the-art ligand systems. This review will highlight the development of Pd(0), Pd(I), and Pd(II) precatalysts that efficiently generate LPd(0) active species with a focus on their activation mechanisms and applications in catalysis. |
|
|
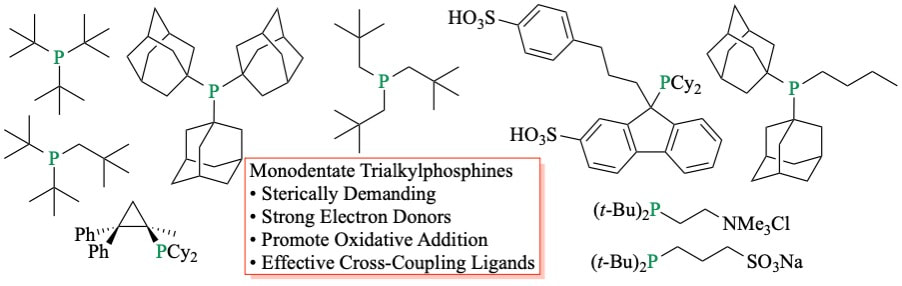
49. "Monodentate Trialkylphosphines as Privileged Ligands in Metal-Catalyzed Cross-Coupling Reactions" Shaughnessy, K. H. Curr. Org. Chem. 2020, 24, 231-264.
Phosphines are widely used ligands in transition metal-catalyzed reactions. Arylphosphines, such as triphenylphosphine, were among the first phosphines to show broad utility in catalysis. Beginning in the late 1990s, sterically demanding and electron-rich trialkylphosphines began to receive attention as supporting ligands. These ligands were found to be particularly effective at promoting oxidative addition in cross-coupling of aryl halides. With electron-rich, sterically demanding ligands, such as tri-tert-butylphosphine, coupling of aryl bromides could be achieved at room temperature. Moreimportantly, the less reactive, but more broadly available, aryl chlorides became accessible substrates. Tri-tert-butylphosphine has become a privileged ligand that has found application in a wide range of late transition-metal catalyzed coupling reactions. This success has led to the use of numerous monodentate trialkylphosphines in cross-coupling reactions. This review will discuss the general properties and features of monodentate trialkylphosphines and their application in cross-coupling reactions of C–X and C–H bonds. |
|
|
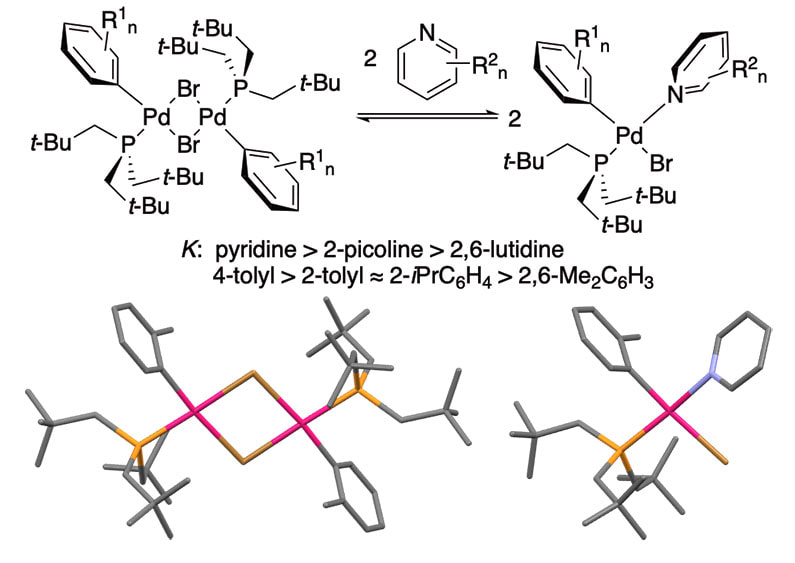
48. "Synthesis, Structural Characterization, and Coordination Chemistry of (Trineopentylphosphine)palladium(aryl)bromide Dimer Complexes ([(Np3P)Pd(Ar)Br]2)," Hu, H.; Vasiliu, M.; Stein, T. H.; Qu, F.; Gerlach, D. L; Dixon, D. A.; Shaughnessy, K. H. Inorg. Chem., 2019, 58, 10299-10313.
A series of [(PNp3)Pd(Ar)Br]2 complexes (PNp3 = trineopentylphosphine, Ar = 4-tolyl, 4-tert-butylphenyl, 2-tolyl, 4-methoxy-2-methylphenyl, 2-isopropylphenyl, and 2,6-dimethylphenyl) were synthesized and structurally characterized by X-ray crystallography and density functional theory optimized structures. The trineopentylphosphine ligand is able to accommodate coordination of other sterically demanding ligands through changes in its conformation. These conformational changes can be seen in changes in percent buried volume of the PNp3 ligand. The binding equilibria of the [(PNp3)Pd(Ar)Br]2 complexes with pyridine derivatives were determined experimentally and analyzed computationally. The binding equilibria are sensitive to the steric demand of the pyridine ligand and less sensitive to the steric demand of the aryl ligand on palladium. In contrast to previous studies, the binding equilibria do not correlate with pyridine basicity. The binding equilibria results are relevant to fundamental ligand coordination steps in cross-coupling reactions, such as Buchwald-Hartwig aminations. |
|
|
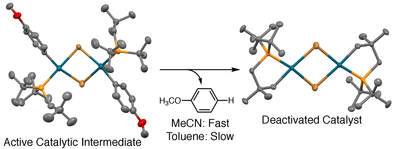
47. "Air-Stable [(R3P)PdCl2]2 Complexes of Neopentylphosphines as Cross-Coupling Precatalysts: Catalytic Application and Mechanism of Catalyst Activation and Deactivation," Barnett, K. L.; Howard, J. R.; Treager, C. J.; Shipley, A. T.; Stullich, R. M.; Qu, F.; Gerlach, D. L.; Shaughnessy, K. H. Organometallics, 2018, 37, 1410-1424.
Air-stable [(R3P)PdCl2]2 complexes with di-tert-butylneopentylphosphine (DTBNpP, 1a) or trineopentylphosphine (TNpP, 1b) ligands have been applied to Suzuki cross-coupling reactions and the mechanism of their conversion to the active LPd(0) catalyst species has been studied. Precatalysts 1a and 1b provide effective catalysts for Suzuki cross-coupling of aryl bromides at room temperature, even when the reactions are performed in air. The precatalyst systems provided much higher activity catalysts in toluene/water compared to acetonitrile/water. Precatalyst loadings could be decreased by a factor of 50 in toluene compared to acetonitrile, while achieving equal or better yields. In acetonitrile/water, ligand metalation to form a [(κ2-P,C-DTBNpP)PdX]2 palladacycle was found to compete with formation of the active Pd(0) species. The palladacycle shows low catalytic activity, so its formation represents a catalyst deactivation pathway. In toluene, clean formation of the active Pd(0) species occurs without competitive palladacycle formation. The improved selectivity to form the active Pd(0) species in toluene appears to account for the higher activity of the precatalysts in toluene/water. |
|
|
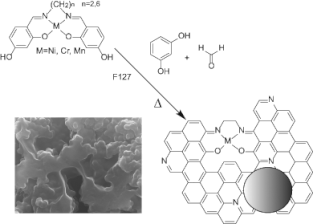
46. "Copolymerization of Transition Metal Salen Complexes and Conversion into Metal Nanoparticles Supported on Hierarchically Porous Carbon Monoliths: A One Pot Synthesis," Kotbagi, T. V.; Shaughnessy, K. H.; LeDoux, C.; Cho, H.; Tay-Agbozo, S.; van Zee, J.; Bakker, M. G. J. Sol-Gel Sci. Technol., 2017, 84, 258-273.
Described is the synthesis of metal (Ni, Cr, and Mn) doped macro/mesoporous carbon monoliths by co- polymerization of transition metal complexes with resorci- nol/formaldehyde and subsequent pyrolysis. Two salen ligands were synthesized via reaction of ethylene diamine (EDA) and 1,6-diaminohexane (DAH) with 2,4-dihydrox- ybenzaldehyde to give 4,4′-((1E,1′E)-(ethane-1,2-diylbis (azanylylidene))bis(methanylylidene))bis(benzene-1,3-diol) (=E-salen) and 4,4′-((1E,1′E)-(hexane-1,6-diylbis(azanyly- lidene))bis(methanylylidene))bis(benzene-1,3-diol) (=D- salen). Their metal salen complexes were prepared. All the samples pyrolyzed at 500 °C had macro and mesopores with a specific surface area ranging from 250–500 m2/g. How- ever, the surface area and porosity of the samples decreased dramatically for the metal containing samples pyrolyzed at 800°C. This is suggested to result from incomplete co- polymerization of the metal salen complexes. The extent of graphitization determined from Raman increased with the pyrolysis temperature. The electrical conductivity was found to increase with the pyrolysis temperature, and fol- lowed the trend Ni>Mn>Cr. The M-E-salen containing samples had significantly lower conductivity than their D- salen counterparts. |
|
|
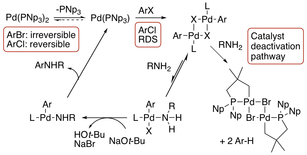
45. "Mechanistic Study of the Role of Substrate Steric Effects and Aniline Inhibition on the Bis(trineopentylphosphine)-palladium(0)-Catalyzed Arylation of Aniline Derivative" Hu, H.; Qu, F.; Gerlach, D. L.; Shaughnessy, K. H. ACS Catal., 2017, 7, 2516-2527.
The mechanism of the bis(trineopentylphosphine)palladium(0) (Pd(PNp3)2)-catalyzed coupling of aryl halides and aniline derivatives was studied in an effort to understand the role of substrate steric effects on the reaction. Prior studies had shown that the rate of Pd/PNp3-catalyzed coupling of aryl bromides and aniline derivatives was largely unaffected by substrate steric demand. The oxidative addition of aryl bromides to Pd(PNp3)2 is found to follow first order kinetics with a rate that is independent of both ligand and aryl halide concentration. Thus, the rate limiting step for oxidative addition of aryl bromides is irreversible ligand dissociation. In the case of aryl chlorides, the oxidative addition rate has a first order dependence on [ArCl] and an inverse dependence on [PNp3] indicating a mechanism involving reversible dissociation of ligand followed by rate limiting oxidative addition. This difference in aryl halide effect was also found for the catalytic coupling reaction. Aryl bromide steric demand does not affect the coupling rate with hindered anilines, whereas the coupling rate of aryl chlorides is negatively affected by substrate steric demand. These results suggest that oxidative addition is rate limiting in the catalytic reaction for aryl chlorides, but that oxidative addition is not rate limiting for aryl bromides. Aniline was found to give significantly slower coupling rates than 2,6-diisopropylaniline for both aryl bromides and chlorides. Aniline promotes the decomposition of the [(PNp3)Pd(Ar)(µ-X)]2 catalytic intermediate to a catalytically inactive palladacycle ([(κ2-P,C-Np2PCH2C(Me2)CH2)Pd(µ-X)]2) through C-H activation of a neopentyl group and elimination of arene. These studies show that the ability of the Pd/PNp3 catalyst system to tolerate steric demand in aryl bromides stems from the fact that the rate limiting step of the catalytic cycle is independent of the concentration and steric demand of aryl bromides. A catalyst deactivation pathway involving ligand metallation has been identified that is promoted by unhindered aniline derivatives. |
|
|
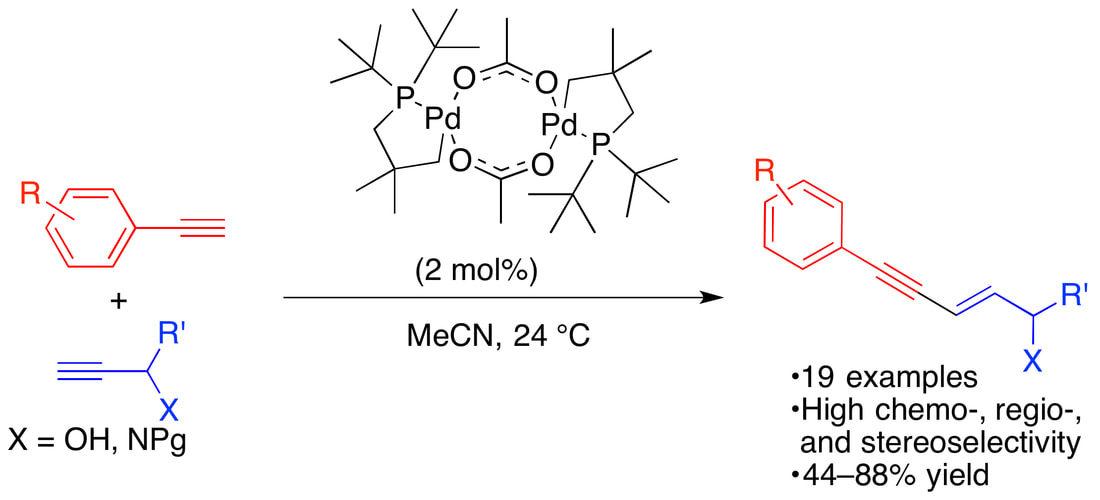
44. "A Trialkylphosphine Derived Palladacycle as a Catalyst in the Selective Cross-Dimerization of Terminal Arylacetylenes with Terminal Propargyl Alcohols and Amides" Lauer, M. G.; Headford, B. R.; Gobble, O. M.; Weyhaupt, M. B.; Gerlach, D. L.; Zeller, M.; Shaughnessy, K. H. ACS Catal., 2016, 6, 5834-5842.
A method for the selective cross-dimerization of terminal aryl alkynes with propargyl alcohols to afford linear E-enynol products is reported. The [Pd(µ- κ2-O,O-OAc)( κ2-C,P-(t-Bu)2PCH2C(Me)2CH2)]2 complex selectively affords E-5-aryl-2-en-4-yn-1-ol products in good yields under mild conditions with high chemo-, regio-, and stereoselectivity. In contrast, previously reported examples of this reaction afford the branched 4-aryl-2-hydroxymethanol-1-buten-3-yne. Propargyl amides are also selectively cross-dimerized, but with lower regioselectivity for the linear enyne. The method has been applied to the synthesis of E-5-phenyl-2-penten-4-yn-1ol, which is a precursor to type 2 diabetes drug candidate NNC 61-4655, in 72% yield from phenyl acetylene and propargyl alcohol. The palladacycle precatalyst reacts with aryl alkynes to afford the first example of a dimeric palladacycle complex with a µ-κ2-C1,C1-bound acetylide ligand. This complex is observed during the catalytic reaction and is a competent precatalyst. |
|
|
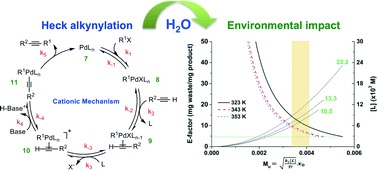
43. "Influence of water on the deprotonation and the ionic mechanisms of a Heck alkynylation and its resultant E-factors," Hu, C.; Shaughnessy, K. H.; Hartman, R. L. React. Chem & Eng. 2016, 1, 65-72. Cover Image
The influence of water on deprotonation and ionic mechanisms of a Heck alkynylation and its resultant E-factors were investigated. Estimation of the Hatta modulus, MH < 0.02, in cationic deprotonation, anionic deprotonation, and the ionic mechanism each separately confirmed an infinitely slow rate of reaction with respect to the diffusive flux within the thin film of the immiscible aqueous–organic interface. As a consequence, intrinsic kinetic expressions for far-equilibrium conditions were derived from first principles for each mechanism. Analyses of Gibbs free energies revealed that water potentially switched the rate-determining steps of cationic and anionic deprotonation to any of oxidative addition of organohalide to form Pd-complex (ΔG‡ = 97.6 kJ mol−1), coordination of the alkyne with the oxidative addition adduct (ΔG‡ = 97.6 kJ mol−1), or ligand substitution to form the cationic Pd-complex (ΔG‡ = 94.9 kJ mol−1). Hydrogen-bonding in the transfer mechanism might account for the switch. Water, in general, was found to influence which step governs each catalytic cycle and the magnitude of its Gibbs free energy. Transformation of the synthesis from batch to continuous-flow was also studied by analyses of E-factors within the thin film. The amount of waste generated, as indicted by estimations of E-factors, was less in continuous-flow operation than in batch when the fastest step of deprotonation (ligand substitution) was infinitely fast with respect to the diffusive flux. The concentration of hydrophilic phosphine ligand was observed to influence mass transport limitations and the E-factor. Increasing ligand concentrations beyond (10.5), (13.3), and (23.2) × 10−3 mol L−1 for reaction temperatures of 353, 343, and 323 K increased the E-factor above its minimum value of 4.7, and it also induced mass-transfer-limitations. The switch from intrinsic to mass-transport-limited kinetics by finite changes in the ligand concentration explains ambiguity when performing aqueous-phase catalyzed Heck alkynylations and possibly multiphase Pd-catalyzed C–C cross-couplings in general. The potential exists to inadvertently mask the reactivity of useful ligands during discovery and to force mass transport limitations during manufacture. Understanding why the E-factor can be minimized is vital to the sustainable discovery and manufacture of fine chemicals, materials, natural products, and pharmaceuticals. |
|
|
42. "Palladium theory of aqueous-phase Heck alkynylations for intensification of discovery and manufacture," Sabio, J. C.; Domier, R. C.; Moore, J. N.; Shaughnessy, K. H.; Hartman, R. L. Chem. Eng. Technol, 2015, 38, 1717-1725.
The influence of water on the catalysis of biphasic Heck alkynylation, a family of palladium-catalyzed carbon-carbon bond formations, was investigated. Kinetic theory derived from Hatta moduli and pseudo-stationary-state approximations discovered that water, in coordination, reductive elimination, and product dissociation reaction steps of the deprotonation catalytic cycle, increases Gibbs energy barriers compared to values previously estimated by density functional theory calculations of purely organic syntheses involving an aryl iodide. On the contrary, water reduces the energy barrier of reductive elimination in the carbopalladation catalytic cycle. Quantum tunneling in proton transfer mechanism might account for the change. The discoveries permitted E-factor predictions that could someday help reduce chemical wastes generated during materials, natural products, and pharmaceutical manufactures. Theoretical groundwork is laid that enables datadriven research in the academic laboratory and data-driven development by the process chemist. |
|
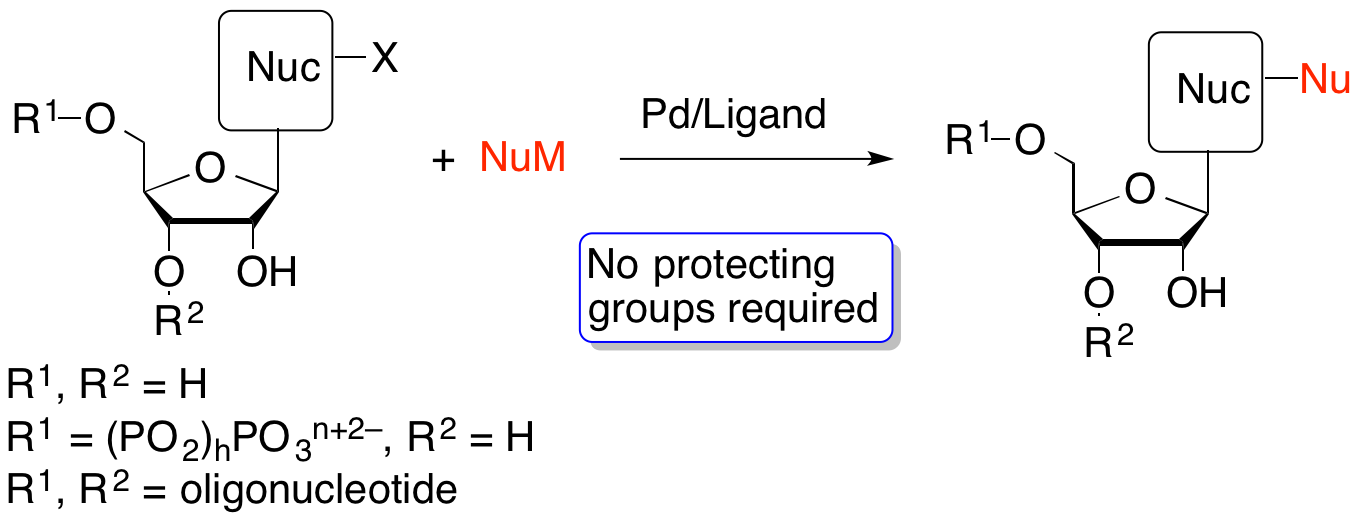
41. "Palladium-Catalyzed Modification of Unprotected Nucleosides, Nucleotides, and Oligonucleotides," Shaughnessy, K. H. Molecules, 2015, 20, 9419-9454.
Synthetic modification of nucleoside structures provides access to molecules of interest as pharmaceuticals, biochemical probes, and models to study diseases. Covalent modification of the purine and pyrimidine bases is an important strategy for the synthesis of these adducts. Palladium-catalyzed cross-coupling is a powerful method to attach groups to the base heterocycles through the formation of new carbon-carbon and carbon-heteroatom bonds. In this review, approaches to palladium-catalyzed modification of unprotected nucleosides, nucleotides, and oligonucleotides are reviewed. Polar reaction media, such as water or polar aprotic solvents, allow reactions to be performed directly on the hydrophilic nucleosides and nucleotides without the need to use protecting groups. Homogeneous aqueous-phase coupling reactions catalyzed by palladium complexes of water-soluble ligands provide a general approach to the synthesis of modified nucleosides, nucleotides, and oligonucleotides. |
|
|
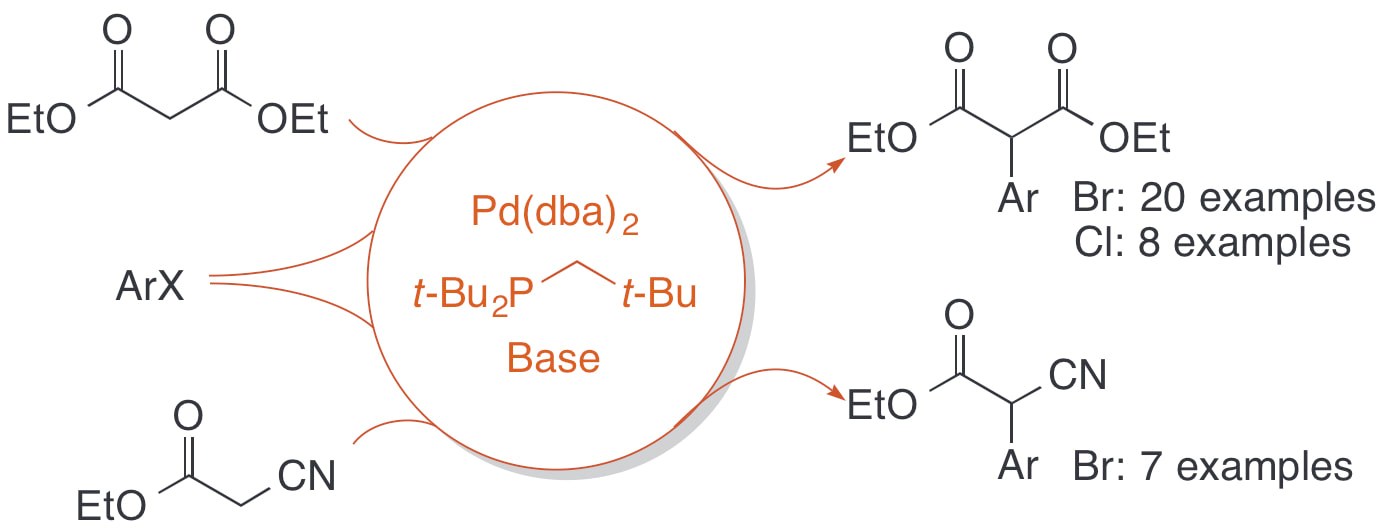
40. "Arylation of diethyl malonate and ethyl cyanoacetate catalyzed by palladium/di-tert-butylneopentyl-phosphine," Semmes, J. G.; Bevans, S. L.; Mullins, C. H.; Shaughnessy, K. H. Tetrahedron Lett., 2015, 56, 3447-3450.
α-Arylated carbonyl derivatives are important structural motifs in many natural products and pharmaceutically active compounds. Although arylation of simple monocarbonyl compounds is a well-established methodology, metal-catalyzed arylation of β-dicarbonyl derivatives is significantly more challenging. The ability of β-dicarbonyl anions to bind to palladium in a κ2-O,O mode, rather than the κ1-C-bound mode required for bond formation, often results in the deactivation of catalyst systems. The C-bound form of the enolate can be favored through the use of sterically demanding ligands. Herein, we report that the sterically demanding di-tert-butylneopentylphosphine (DTBNpP) ligand in combination with Pd(dba)2 provides an effective catalyst for the coupling of aryl bromides and chlorides with diethyl malonate. The Pd/DTBNpP system also catalyzes the coupling of aryl bromides with ethyl cyanoacetate. |
|
|
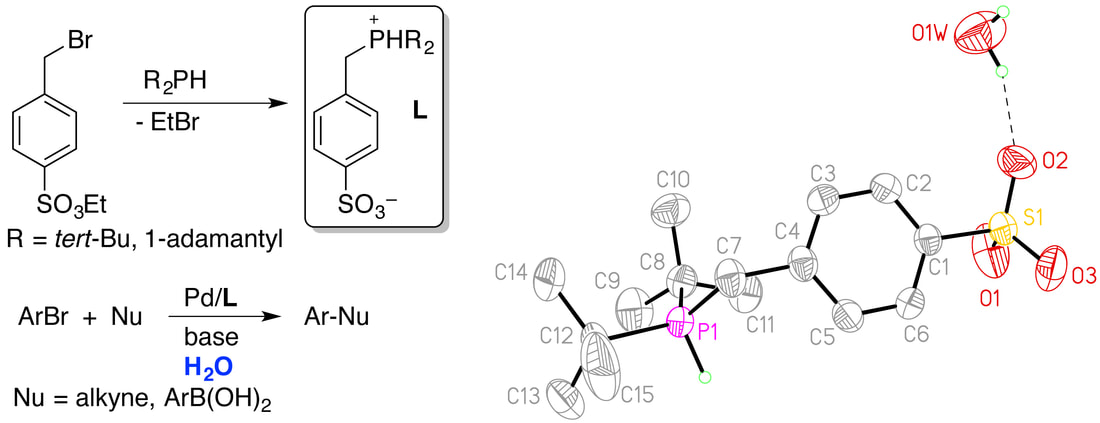
39. "Synthesis of 4-Sulfonatobenzylphosphines and Their Application in Aqueous-Phase Palladium-Catalyzed Cross-Coupling," Moore, J. N.; Laskay, N. M.; Duque, K. S.; Kelley, S. P.; Rogers, R. D.; Shaughnessy, K. H. J. Organomet. Chem. 2015, 777, 16-24.
Aqueous-biphasic catalysis offers the potential for safer and more environmentally sustainable synthetic processes. In addition, hydrophilic supporting ligands allow homogeneous catalysts to be readily separated from organic products and potentially reused. The synthesis of two new water-soluble ligand precursors, di-tert-butyl(4-sulfonatobenzyl)phosphonium and di-1-adamantyl(4-sulfonatobenzyl)phosphonium, are reported. The air-stable, zwitterionic phosphonium salts were prepared by the reaction of dialkylphosphines with ethyl 4-bromomethylbenzenesulfonate, which results in a one-pot alkylation followed by deprotection of the ethyl sulfonate. This methodology provides an operationally simpler route to sulfonated benzylphosphines than electrophilic sulfonation. The new phosphine ligands were applied to aqueous-phase Suzuki and Sonogashira couplings of aryl bromides. |
|
|
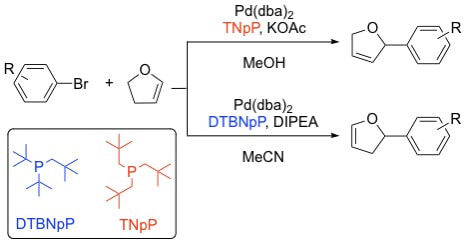
38. "Controlling Olefin Isomerization in the Heck Reaction with Neopentyl Phosphine Ligands," Lauer, M. G.; Thompson, M. K.; Shaughnessy, K. H. J. Org. Chem., 2014, 79, 10837-10848.
Highlighted in: "Highlights from the Literature," Org. Proc. Res. Dev., 2015, 19, 250-258. The use of neopentyl phosphine ligands were examined in the coupling of aryl bromides with alkenes. Di-tert-butylneopentylphosphine (DTBNpP) was found to catalyze Heck couplings with aryl bromides at ambient temperature. In the Heck coupling of cyclic alkenes, the degree of alkene isomerization was found to be controlled by the choice of ligand with DTBNpP promoting isomerization to a much greater extent than trineopentylphosphine (TNpP). Under optimal conditions, DTBNpP provides high selectivity for 2-aryl-2,3-dihydrofuran in the arylation of 2,3-dihydrofuran, whereas TNpP provided high selectivity for the isomeric 2-aryl-2,5-dihydrofuran. A similar complimentary product selectivity is seen in the Heck coupling of cyclopentene. Heck coupling of 2-bromophenols or 2-bromoanilides with 2,3-dihydrofurans affords 2,5-epoxybenzoxepin and 2,5-epoxybenzazepins, respectively. |
|
|
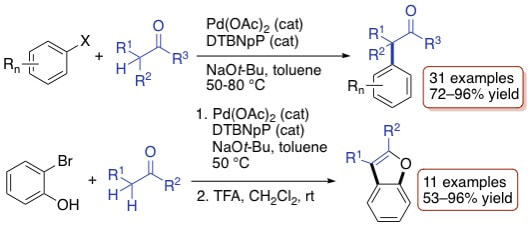
37. "Di-tert-butylneopentylphosphine (DTBNpP): An Efficient Ligand in the Palladium-Catalyzed α-Arylation of Ketones," Raders, S. M.; Jones, J. M.; Semmes, J. G.; Kelley, S. P.; Rogers, R. D; Shaughnessy, K. H. Eur. J. Org. Chem. 2014, 7395-7404.
Di-tert-butylneopentylphosphine (DTBNpP) and palladium(II) acetate provide an efficient catalytic system for the a-arylation of ketones. Aryl bromides were coupled with ketones using 0.25-0.5 mol % Pd(OAc)2/DTBNpP in toluene at 50 °C, whereas aryl chlorides required a higher catalyst loading (0.5-2.0 mol %) and a higher temperature (80 °C). Coupling of 2-bromphenol with ketones using the Pd/DTBNpP system provides an efficient route for the synthesis of benzofurans. |
|
|
36. "Palladium-catalyzed ortho-halogenation of diaryl oxime ethers," Bhattarai, B. T. Adhikari, S.; Kimball, E. A.; Moore, J. N.; Shaughnessy, K. H.; Snowden, T. S.; Fronczek, F. R.; Dolliver, D. D. Tetrahedron Lett. 2014, 55, 4801-4806.
The palladium-catalyzed ortho monobromination and iodination of single E or Z geometric isomers of substituted diaryl ketoxime ethers [Ar1C(Ar2) = N–OCH3] have been accomplished in good yield (64–82% isolated yield). A minor amount of di-ortho-halogenation side product is produced in this reaction (0–10%). The aromatic ring that is halogenated is the one ‘trans’ to the OCH3 group, indicating that there is no isomerization about the C@N bond during the reaction. Further, the site of functionalization is presumably controlled by the lone pair electrons on nitrogen. This study is the first to demonstrate the ability of the geometry of the oxime ether group to target the functionalization of only one aromatic ring when there are two aromatic rings directly attached to the carbon of the C–N bond of the oxime ether. Electron-withdrawing substituents on either ring decrease reaction rate and diminish the amount of di-ortho-bromination side product. |

|
|
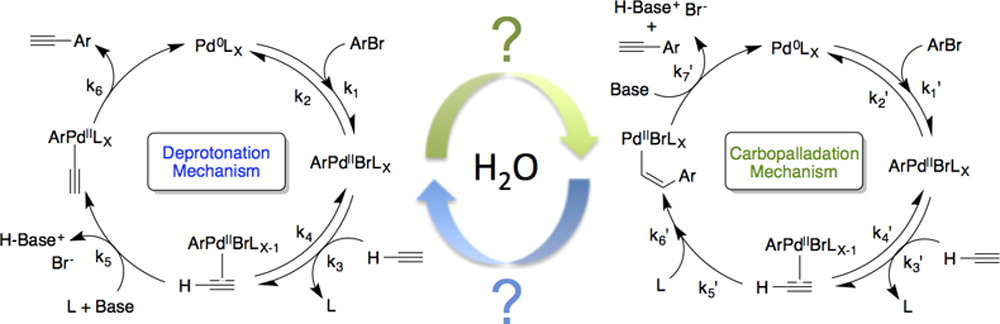
35. "Kinetic Analysis of Aqueous-Phase Pd-Catalyzed, Cu-Free Direct Arylation of Terminal Alkynes Using a Hydrophilic Ligand," Moore, J. N.; Domier, R. C.; Shaughnessy, K. H.; Hartman, R. L. Org. Proc. Res. Dev., 2013, 17, 1262-1271.
The engineering of reaction mixtures that ensure the solubility of inorganic salt byproducts without compromising the reactivity is a grand challenge for continuous flow manufacturing in upstream pharmaceuticals and fine chemicals process development. Aqueous cross-coupling reactions are possible solutions. We report the study of an aqueous phase Pd-catalyzed Cufree direct arylation of an alkyne using a hydrophilic ligand towards understanding the role of water on the cross-coupling kinetics. Kinetic analyses of theoretically estimated molar flux rates and the measured reaction kinetics reveal a transition from mass transfer to kinetically controlled deprotonation and carbopalladation mechanisms. Interfacial contact areas of immiscible aqueous−organic phases control the crossover from the mass-transfer-limited to the reaction-rate-limited regime. Highly agitated batch reactors and multiphase capillary flow reactors are needed to overcome the mass transport limitations and, thus, discover the transition from the apparent reaction kinetics to the true reaction kinetics. Comparison of previously reported Density Functional Theory calculations with experimentally measured activation energies, ranging from 14.8 to 20.0 kcal/mol, elucidates the theoretical possibility of designing the aqueous phase C−C cross-coupling reaction with similar reactivity as purely organic reactions. Although ambiguity remains concerning which reaction step is rate-determining in either the deprotonation or the carbopalladation mechanism, our discovery undergirds that one mechanism or the other could dominate in aqueous designed direct arylations. |
|
|
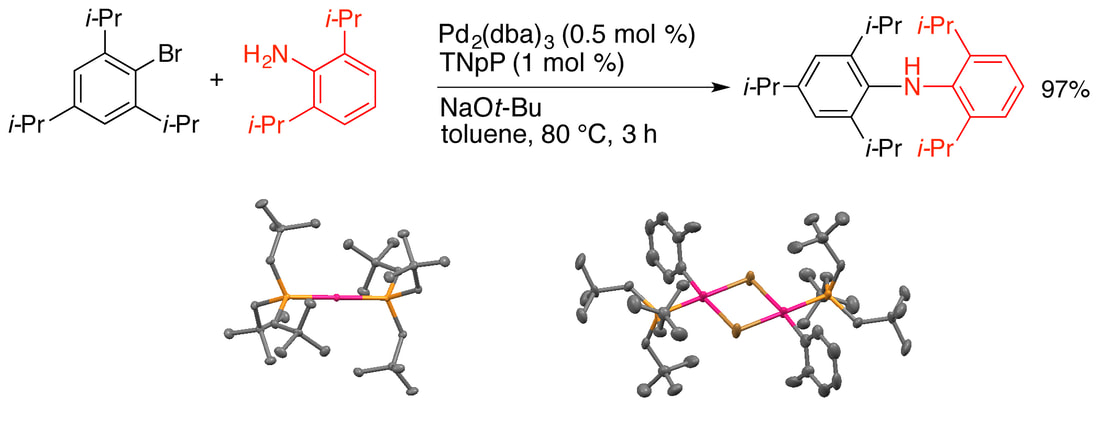
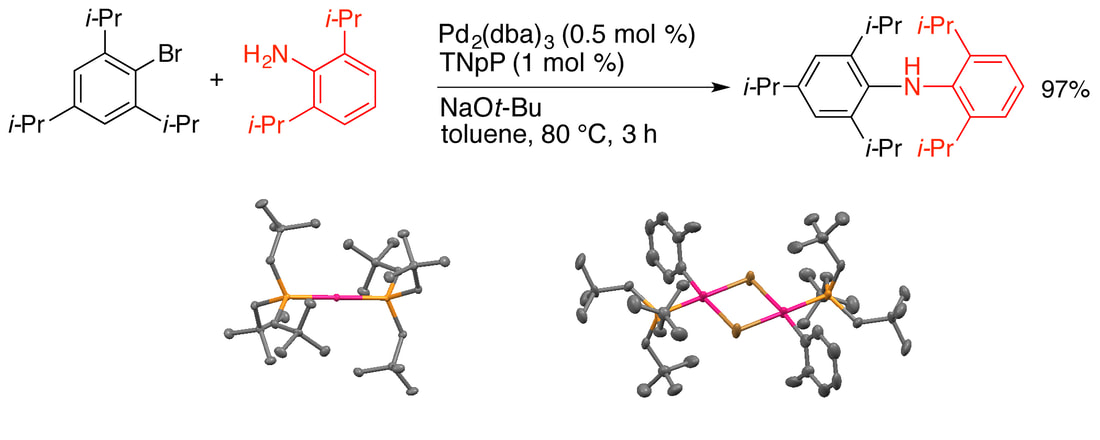
34. " Trineopentylphosphine: A Conformationally Flexible Ligand for the Coupling of Sterically Demanding Substrates in the Buchwald-Hartwig Amination and Suzuki-Miyaura Reaction," Raders, S. M.; Moore, J. N.; Parks, J. K.; Miller, A. D.; Leißing, T. M.; Kelley, S. P.; Rogers, R. D.; Shaughnessy, K. H. J. Org. Chem., 2013, 78, 4649-4664. Featured Article
Trineopentylphosphine (TNpP) in combination with palladium provides a highly effective catalyst for the Buchwald-Hartwig coupling of sterically demanding aryl bromides and chlorides with sterically hindered aniline derivatives. Excellent yields are obtained even when both substrates include 2,6-diisopropyl substituents. Notably, the reaction rate is inversely related to the steric demand of the substrates. X-Ray crystallographic structures of Pd(TNpP)2, [Pd(4-t-Bu-C6H4)(TNpP)(µ-Br)]2, and [Pd(2-Me-C6H4)(TNpP)(µ-Br)]2 are reported. These structures suggest that the conformational flexibility of the TNpP ligand plays a key role in allowing the catalyst to couple hindered substrates. The Pd/TNpP system also shows good activity for the Suzuki coupling of hindered aryl bromides. |
|
|
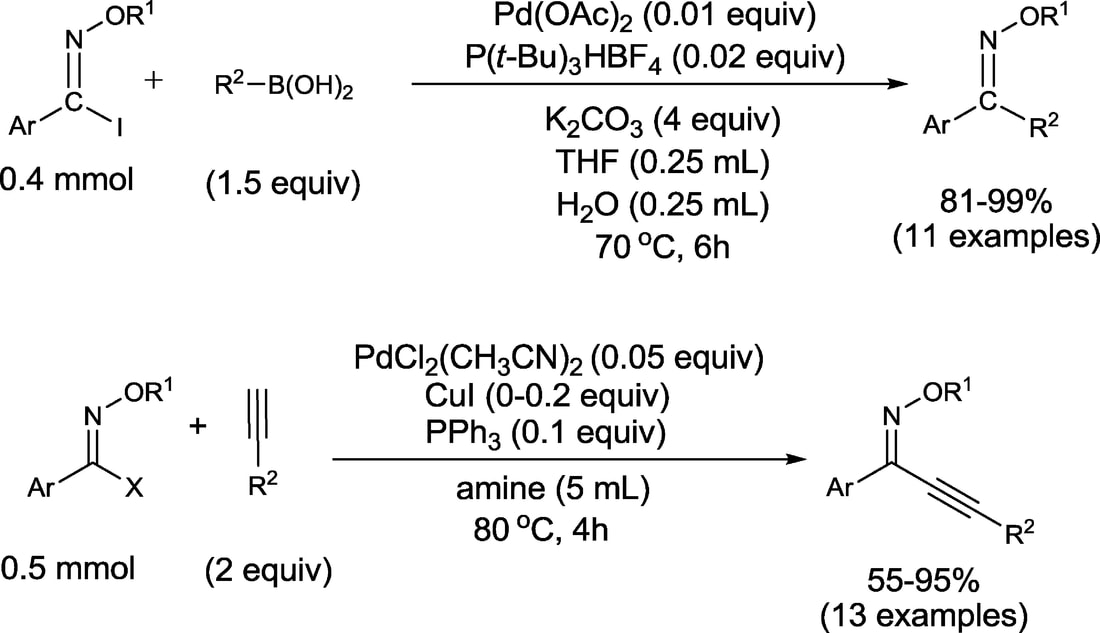
33. "Stereospecific Suzuki, Sonogashira, and Negishi Coupling Reactions of N-Alkoxyimidoyl Iodides and Bromides," Dolliver, D. D.; Bhattarai, B.; Pandey, A.; Lanier, M. L.; Bordelon, A. S.; Adhikari, S.; Dinser, J. A.; Flowers, P. F.; Wills, V. S.; Schneider, C. L.; Shaughnessy, K. H.; Moore, J. N.; Raders, S. M.; Snowden, T. S.; McKim, A. S.; Fronczek, F. R. J. Org. Chem., 2013, 78, 3676-3687.
A high-yielding stereospecific route to the synthesis of single geometric isomers of diaryl oxime ethers through Suzuki coupling of N-alkoxyimidoyl iodides is described. This reaction occurs with complete retention of the imidoyl halide geometry to give single E or Z isomers of diaryl oxime ethers. The Sonogashira coupling of N-alkoxyimidoyl iodides and bromides with a wide variety of terminal alkynes to afford single geometric isomers of aryl alkynyl oxime ethers has also been developed. Several of these reactions proceed through copper-free conditions. The Negishi coupling of N-alkoxyimidoyl halides is introduced. The E and Z configurations of nine Suzuki-coupling products and two Sonogashira-coupling products were confirmed by X-ray crystallography. |
|
|
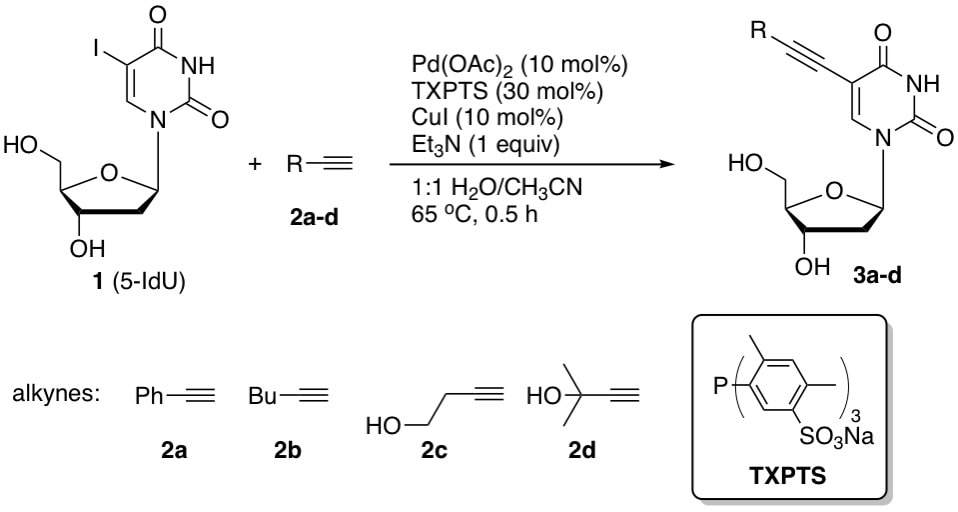
32. "Aqueous-phase Sonogashira alkynylation to synthesize 5-substituted pyrimidine and 8-substituted purine nucleosides," Cho, J. H.; Shaughnessy, K. H. Curr. Prot. Nucleic Acid Chem. 2012, 49, 1.27.1-10.
In this unit, an efficient method for the synthesis of alkyne-modified nucleosides in an aqueous solvent system is described. The method allows direct palladium-catalyzed alkynylation of readily available unprotected 8-bromo-2'-deoxyguanosine (8-BrdG), 8-bromo-2'-deoxyadenosene (8-BrdA) 8-bromoadenosine (8-BrA), and 5-iodo-2'-deoxyuridine (5-IdU) precursors. The optimal catalyst is derived from palladium acetate, tri-(2,4-dimethyl-5-sulfonatophenyl)phosphane (TXPTS), and CuI. |
|
|
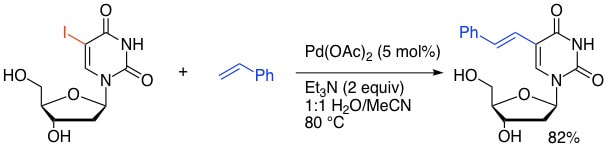
31. "Aqueous-Phase Heck Coupling of 5-Iodouridine and Alkenes under Phosphine-Free Conditions." Cho, J. H.; Shaughnessy, K. H. Synlett 2011, 2963-2966.
Palladium-catalyzed Heck couplings between 5-iodouridine and alkenes were carried out in aqueous media in the presence of triethylamine under phosphine-free conditions to give the desired products in high yields. In the absence of phosphine ligand, triethylamine appears to play a critical role in both generating the active catalyst species and serving as the base in the reaction. |
|
|
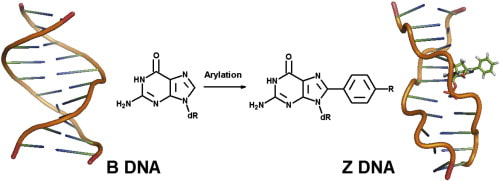
30. "The Conformational Effect of para-Substituted C8-Arylguanine Adducts on the B/Z/-DNA Equilibrium," Vongsutilers, V.; Phillips, D. J.; Train, B. C.; McKelvey, G. R.; Thomsen, N. M.; Shaughnessy, K. H.; Lewis, J. P.; Gannett, P. M. Biophys. Chem., 2011, 154, 41-48.
The B form of DNA exists in equilibrium with the Z form and is mainly affected by sequence, electrostatic interactions, and steric effects. C8-purine substitution shifts the equilibrium toward the Z form though how this interaction overcomes the unfavorable electrostatic interactions and decrease in stacking in the Z form has not been determined. Here, a series of C8-arylguanine derivatives, bearing a para-substituent were prepared and the B/Z equilibrium determined. B/Z ratios were measured by CD and conformational effects of the aryl substitution determined by NMR spectroscopy and molecular modeling. The para-substituent was found to have a significant effect on the B/Z DNA equilibrium caused by altering base-pair stacking of the B form and modifying the hydration/ion shell of the B form. A unique melting temperature versus salt concentration was observed and provides evidence relevant to the mechanism of B/Z conformational interconversion. |
|
|
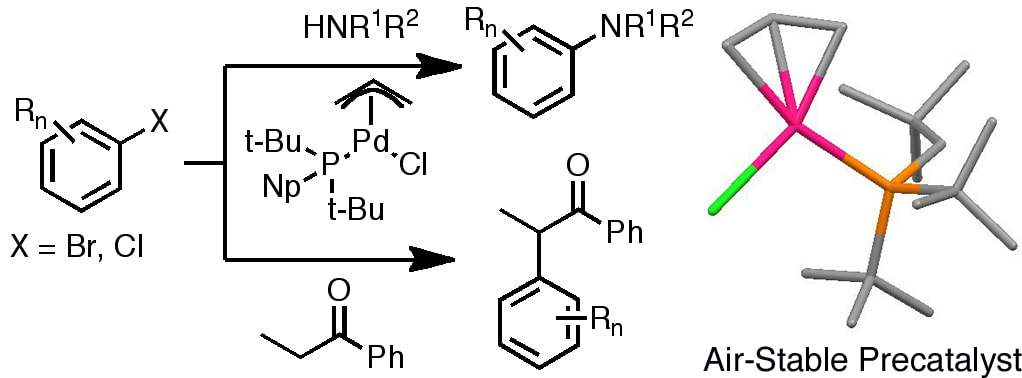
29. "Synthesis and X-ray Structure Determination of Highly Active Pd(II), Pd(I), and Pd(0) Complexes of Di(tert-butyl)neopentylphosphine (DTBNpP) in the Arylation of Amines and Ketones,"Hill, L. L.; Crowell, J. L.; Tutwiler, S. L., Massie, N. L., Hines, C. C.; Griffin, S. T.; Rogers, R. D.; Shaughnessy, K. H.; Grasa, G. A.; Johansson Seechurn, C. C. C.; Li, H.; Colacot, T. J.; Chou, J.; Woltermann, C. J. J. Org. Chem. 2010, 75, 6477–6488.
The air-stable complex Pd(η3-allyl)(DTBNpP)Cl (DTBNpP = di-(tert-butyl)neopentylphosphine) serves as a highly efficient precatalyst for the arylation of amines and enolates using aryl bromides and chlorides under mild conditions with yields ranging from 74-98%. Amination reactions of aryl bromides were carried using 1-2 mol% Pd(η3-allyl)(DTBNpP)Cl at 23-50 °C without the need to exclude oxygen or moisture. The C-N coupling of the aryl chlorides occurred at relatively lower temperature (80-100 °C) and catalyst loading (1 mol%) using the Pd(η3-allyl)(DTBNpP)Cl precatalyst than the catalyst generated in situ from DTBNpP and Pd2(dba)3 (100-140 °C, 2-5 mol% Pd). Other Pd(DTBNpP)2-based complexes, (Pd(DTBNpP)2 and Pd(DTBNpP)2Cl2) were ineffective precatalysts under identical conditions for the amination reactions. Both Pd(DTBNpP)2 and Pd(DTBNpP)2Cl2 precatalysts gave nearly quantitative conversions to the product in the a-arylation of p-chlorotoluene and p-bromoanisole with propiophenone at a substrate/catalyst loading of 100/1, however. At lower substrate/catalyst loading (1000/1), the conversions were lower, but comparable to that of Pd(t-Bu3P)2. In many cases, the tri-tert-butylphosphine (TTBP) based Pd(I) dimer, [Pd(µ-Br)(TTBP)]2 stood out to be the most reactive catalyst under identical conditions for the enolate arylation. Interestingly, the air stable Pd(I) dimer, Pd2(DTBNpP)2(µ-Cl)(µ-allyl) was less active in comparison to [Pd(µ-Br)(TTBP)]2 and Pd(η3-allyl)(DTBNpP)Cl. The X-ray crystal structures of Pd(η3-allyl)(DTBNpP)Cl, Pd(DTBNpP)2Cl2, Pd(DTBNpP)2 and Pd2(DTBNpP)2(µ-Cl)(µ-allyl) are reported in this paper along with initial studies on the catalyst activation of the Pd(η3-allyl)(DTBNpP)Cl precatalyst. |
|
|
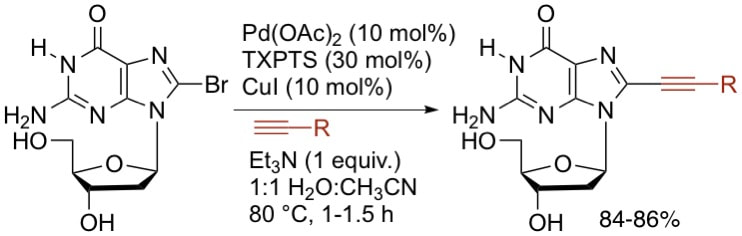
28. "Efficient Sonogashira Coupling of Unprotected Halonucleosides in Aqueous Solvents Using Water-Soluble Palladium Catalysts," Cho, J. H.; Prickett, C. D.; Shaughnessy, K. H. Eur. J. Org. Chem. 2010, 3678-3683.
A variety of applications of C-alkynylated nucleosides has prompted the continuing development of efficient synthetic methods for their preparation. We have developed an efficient and environmentally benign Sonogashira coupling reaction for alkynylation of unprotected halonucleosides in an aqueous solvent. The combination of Pd(OAc)2, CuI, and TXPTS (trisodium tri(2,4-dimethyl-5-sulfonatophenyl)phosphine) provided a highly active catalyst for the alkynylation of 8-bromopurines and 5-iodouridine in 1:1 H2O/CH3CN in yields ranging from 42-98%. |
|
|
27. "Prediction of Reliable Metal–PH3 Bond Energies for Ni, Pd, and Pt in the 0 and +2 Oxidation States" Craciun, R.; Vincent, A. J.; Shaughnessy, K. H.; Dixon, D. A. Inorg. Chem., 2010, 49, 5546-5553.
"Correction to Prediction of Reliable Metal–PH3 Bond Energies for Ni, Pd, and Pt in the 0 and +2 Oxidation States." Craciun, R.; Vincent, A. J.; Shaughnessy, K. H.; Dixon, D. A., Inorg. Chem. 2011, 50, 5307. Phosphine-based catalysts play an important role in many metal-catalyzed carbon-carbon bond formation reactions yet reliable values of their bond energies are not available. We have been studying homogeneous catalysts consisting of a phosphine bonded to a Pt, Pd or Ni. High level electronic structure calculations at the CCSD(T)/complete basis set level were used to predict the M-PH3 bond energy (BE) for the 0 and + 2 oxidation states for M = Ni, Pd, and Pt. A wide range of exchange-correlation functionals were also evaluated to assess the performance of density theory for these important bond energies. These bond energies can then be used, for example, in the design of new catalyst systems. |

|
|
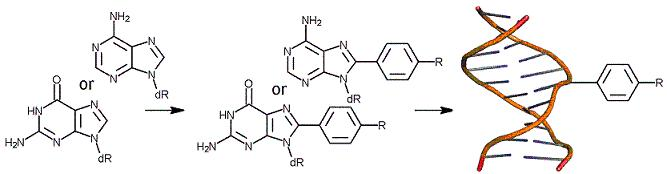
26. "A General Synthesis of C8-Arylpurine Phosphoramidites," Vongsutilers, V.; Daft, J. R.; Shaughnessy, K. H.; Gannett, P. M. Molecules, 2009, 14, 3339-3352.
A general scheme for the synthesis of C8-arylpurine phosphoramidites has been developed. C8-Arylation of C8-bromo-2′-deoxyguanosine is the key step and has been achieved through the use of a Suzuki coupling. Since the coupling reaction is conducted under aqueous conditions, it is unnecessary to protect and then deprotect the hydroxyl groups, thus saving several steps and improving overall yields. Once the C8-arylgroup is introduced, the glycosidic bond becomes very sensitive to acid catalyzed cleavage. Protection of the amino groups as the corresponding N,N-dimethylformamidine derivative improves stability of the derivatives. Synthetic C8-arylpurines were successfully used to prepare synthetic oligonucleotides. |
|
|
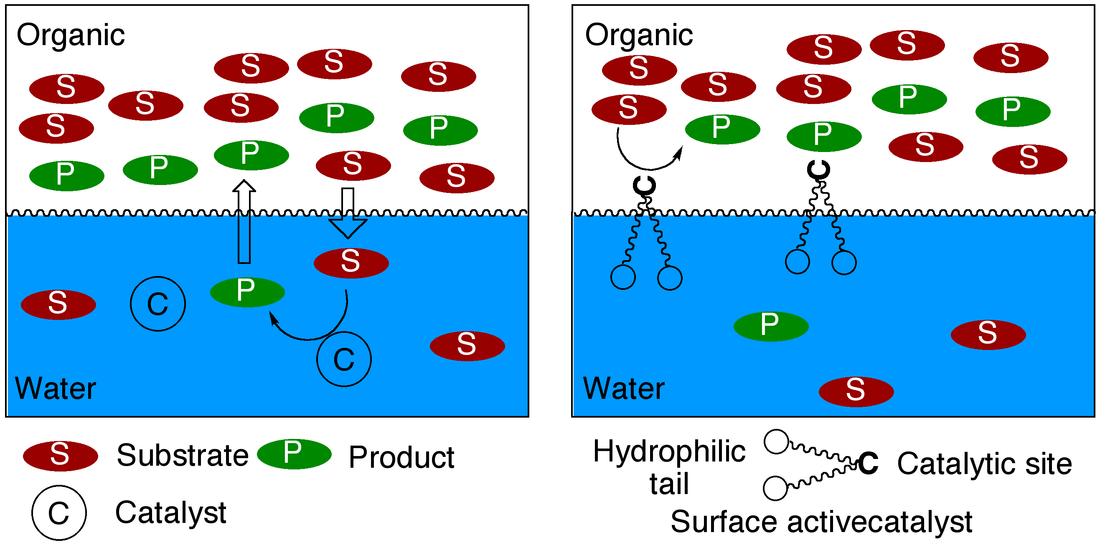
25. "Hydrophilic Ligands and Their Application in Aqueous-Phase Metal-Catalyzed Reactions," Shaughnessy, K. H. Chem. Rev, 2009, 109 ,643-710.
|
|
|
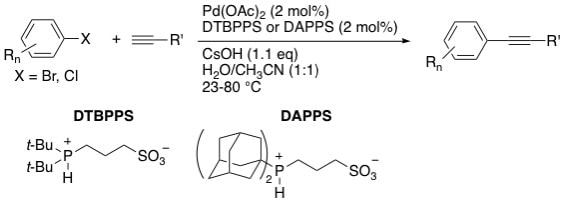
24. "Sterically-Demanding, Zwitterionic Trialkylphosphonium Sulfonates as Air-Stable Ligand Precursors for Efficient Palladium-Catalyzed Cross-Couplings of Aryl Bromides and Chlorides," Brown, W. S.; Boykin, D. D.; Sonnier, M. Q., Jr.; Clark, W. D.; Brown, F. V.; Shaughnessy, K. H. Synthesis, 2008, 1965-1970.
• Invited manuscript 3-(Di-tert-butylphosphonium)propane sulfonate (DTBPPS) and 3-(diadamantylphosphonium)propane sulfonate (DAPPS) are air-stable pre-ligands for aqueous-phase Pd-catalyzed cross-coupling reactions. Both DTBPPS and DAPPS were found to give active catalysts for the Sonogashira coupling of aryl bromides at room temperature and 4-chloroanisole at 80 °C. These ligands also gave effective catalysts for the aqueous-phase Suzuki coupling of aryl bromides at room temperature. |
|
|
23. "Neopentylphosphines as effective ligands in palladium-catalyzed cross-couplings of aryl bromides and chlorides" Hill, L. L.; Smith, J. M.; Brown, W. S.; Moore, L. R.; Guevara, P. J.; Pair, E. S.; Porter, J. E.; Chou, J.; Woltermann, C. J.; Craciun, R.; Dixon, D. A.; Shaughnessy, K. H. Tetrahedron, 2008, 64, 6920-6934.
• Invited Symposium in Print manuscript The use of neopentylphosphine ligands in the palladium-catalyzed Suzuki, Sonogashira, Heck and Hartwig-Buchwald couplings of aryl bromides and chlorides are reported. Di-tert-butylneopentylphosphine (DTBNpP) provided highly active catalysts for the coupling of aryl bromides at mild temperatures. Trineopentylphosphine, an air-stable trialkylphosphine, gave inactive catalysts at room temperature, but showed good activity in the H-B amination of aryl chlorides at elevated temperatures. |
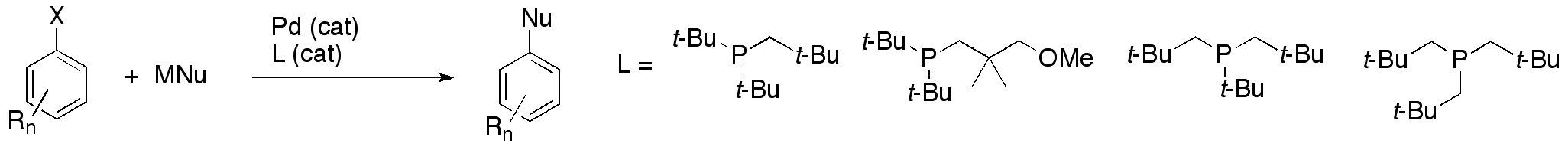
|
|
22. "Sterically Demanding, Sulfonated, Triarylphosphines: Application to Palladium-Catalyzed Cross-Coupling, Steric and Electronic Properties, and Coordination Chemistry," Moore, L. R.; Western, E. C.; Craciun, R.; Spruell, J. M.; Dixon, D. A.; O'Halloran, K. P.; Shaughnessy, K. H. Organometallics, 2008, 27, 576-593.
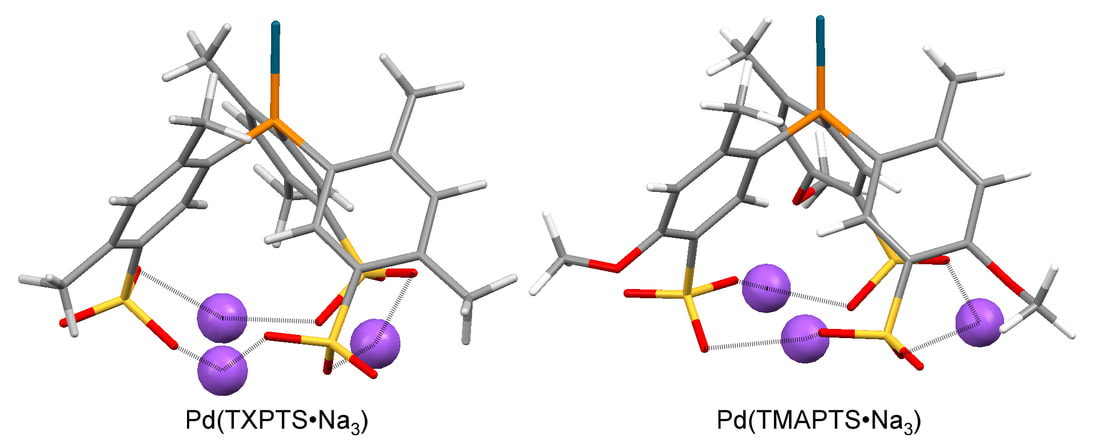
Tri(2,4-dimethyl-5-sulfonatophenyl)phosphine trisodium (TXPTS•Na3) and tri(4-methoxy-2-methyl-5-sulfonatophenyl)phosphine trisodium (TMAPTS•Na3) both provide more active catalysts for Suzuki and Sonogashira couplings of aryl bromides in aqueous solvents than tri(3-sulfonatophenyl)phosphine trisodium (TPPTS•Na3). In the Heck coupling, TXPTS•Na3 provides the most effective catalyst system. Cone angles determined from DFT-optimized structures show that both TXPTS•Na3 (206°) and TMAPTS•Na3 (208°) are significantly larger than TPPTS•Na3 (165°). The identity of the counterion had a significant effect on the calculated cone angles for these ligands. The electronic properties of these ligands determined by the CO stretching frequencies of trans-RhL2(Cl)CO complexes were identical, although calculated electronic parameters suggest subtle differences between these ligands. Similar to TPPTS•Na3, both TXPTS•Na3 and TMAPTS•Na3 react with Pd(OAc)2 in aqueous solvents to give LnPd0 complexes and the corresponding phosphine oxide. The reduction of palladium(II) by TXPTS•Na3 is significantly slower than is seen with TMAPTS•Na3 or TPPTS•Na3 at room temperature. Evidence of palladacycle complexes derived from TXPTS•Na3 and TMAPTS•Na3 by activation of an ortho-methyl substituent was also observed in ligand coordination studies and under catalytic reaction conditions. |
|
|
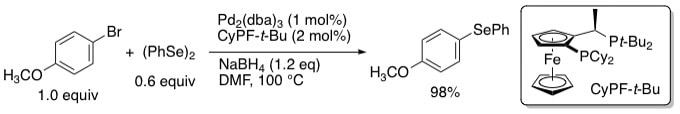
21. "A Selective and Tin-Free Pd-Catalyzed Phenylselenylation of Aryl Bromides" Cho, J. H.; Brown, F. V.; Shaughnessy, K. H. Main Group Chem. 2007, 7, 210-214.
• Invited symposium paper The first selective palladium-catalyzed phenylselenylation of aryl bromides using a tin-free phenylselenolate source is reported. High selectivity for the desired asymmetric diaryl selenide is achieved using a catalyst derived from palladium and a Josiphos-type ligand with (PhSe)2/NaBH4 as the phenylselenylating agent. Aryl bromides are phenylselenylated at 100 °C using low catalyst loadings to give the desired asymmetric diaryl selenides in high yield and >95% selectivity. Phenylselenoborane adducts formed by the reduction of diphenyl diselenide with sodium borohydride are more selective phenylselenylating reagents than sodium phenylselenolate. |
|
|
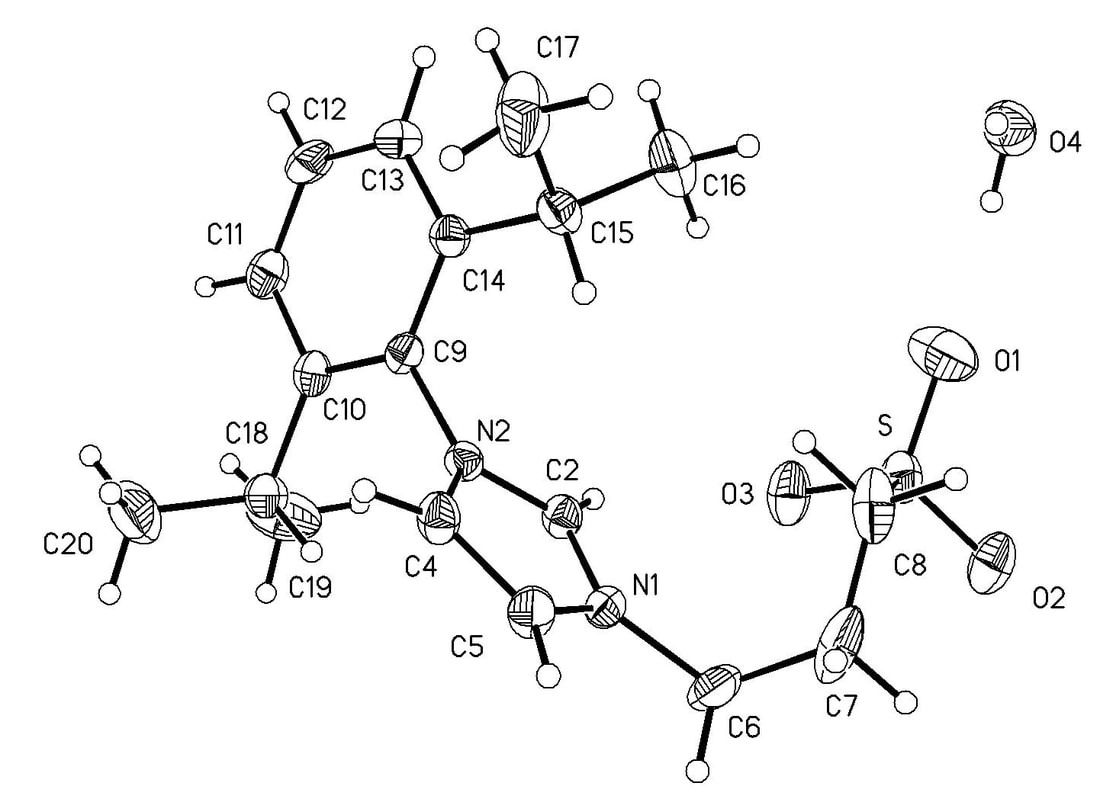
20. "Synthesis and Characterization of Water-Soluble Silver and Palladium Imidazol-2-ylidene Complexes with Non-Coordinated Anionic Substituents, " Moore, L. R.; Cooks, S. M; Anderson, M. S.; Schanz, H.-J.; Griffin, S. T.; Rogers, R. D.; Kirk, M. C; Shaughnessy, K. H. Organometallics, 2006, 25, 5151-5158.
Zwitterionic imidazolium salts have been synthesized bearing alkylsulfonate and alkylcarboxylate substituents and used as precursors to water-soluble metal-carbene complexes. Reaction of the zwitterionic imidazolium compounds with Ag2O gave bis(imidazol-2-ylidene)silver complexes. These compounds have been characterized spectroscopically and by electrospray mass spectrometry. A DMSO solvate of bis[1-(2,6-diisopropylphenyl)-3-(3-sulfonatopropyl)imidazol-2-ylidene]silver sodium salt has been structurally characterized. In the solid state, this complex exists as a coordination polymer in which the sodium ions bridge the sulfonate groups from two bis(imidazol-2-ylidene)silver moieties. Diiodo-bis[1-mesityl-3-(3-sulfonatopropyl)imidazol-2-ylidene]palladium disodium salt has also been prepared in low yield and characterized by NMR spectroscopy and electrospray mass spectrometry. |
|
|
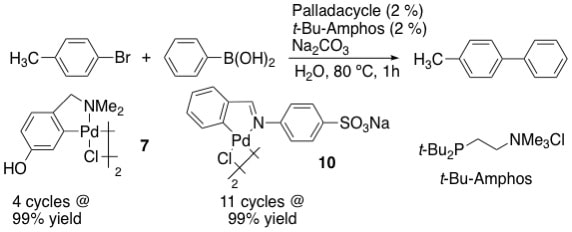
19. "Water-Soluble Palladacycles as Precursors to Highly Recyclable Catalysts for the Suzuki Coupling of Aryl Bromides in Aqueous Solvents," Huang, R. and Shaughnessy, K. H. Organometallics, 2006, 25, 4105-4112.
• #2 most accessed article for July-September 2006 A family of water-soluble palladacycles was prepared from benzyl amine or benzaldehyde imine ligands bearing hydrophilic functional groups. The palladacycles derived from N,N-dimethyl-p-hydroxybenzylamine (7) and sodium 4-(N-benzylideneamino)benzenesulfonate (10) gave active catalysts for the Suzuki coupling of aryl bromides and activated aryl chlorides in combination with (2-di-t-butylphosphinoethyl)trimethylammonium chloride (t-Bu-Amphos). The catalyst derived from 10/t-Bu-Amphos could be used for 12 reaction cycles in the Suzuki coupling of 4-bromotoluene at 80 ºC before a significant loss of catalyst activity was observed. |
|
|
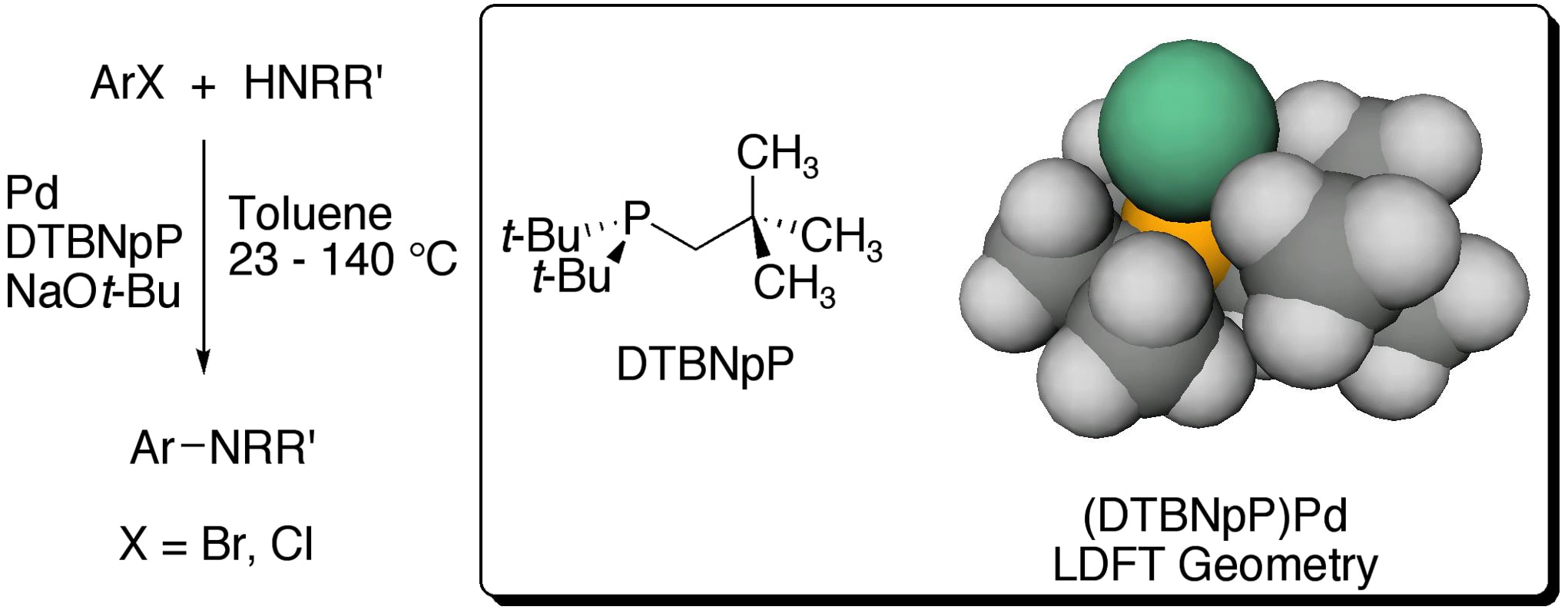
18. "Bulky Alkylphosphines with Neopentyl Substituents as Ligands in the Amination of Aryl Bromides and Chlorides," Hill, L. L.; Moore, L. R.; Huang, R.; Craciun, R.; Vincent, A. J.; Dixon, D. A.; Chou, J.; Woltermann, C. J.; Shaughnessy, K. H. J. Org. Chem., 2006, 71, 5117-5125.
Di(tert-butyl)neopentylphosphine (DTBNpP) in combination with palladium sources provided catalysts with comparable or better activity for the Hartwig-Buchwald amination of aryl bromides than tri(t-butyl)phosphine (TTBP) under mild conditions. DTBNpP also provided effective catalysts for amination reactions of aryl chlorides at elevated temperatures. Further replacement of tert-butyl groups with neopentyl substituents resulted in less effective ligands for amination reactions. Computationally derived cone angles showed that replacement of a tert-butyl group with a neopentyl group significantly increased the cone angle of the phosphine. The larger cone angle of DTBNpP than TTBP appears to correlate with the higher activity of catalysts derived from DTBNpP in the amination of aryl bromides. TTBP is a stronger electron donor than DTBNpP, which may explain the higher activity for TTBP-derived catalysts towards aryl chlorides. |
|
|
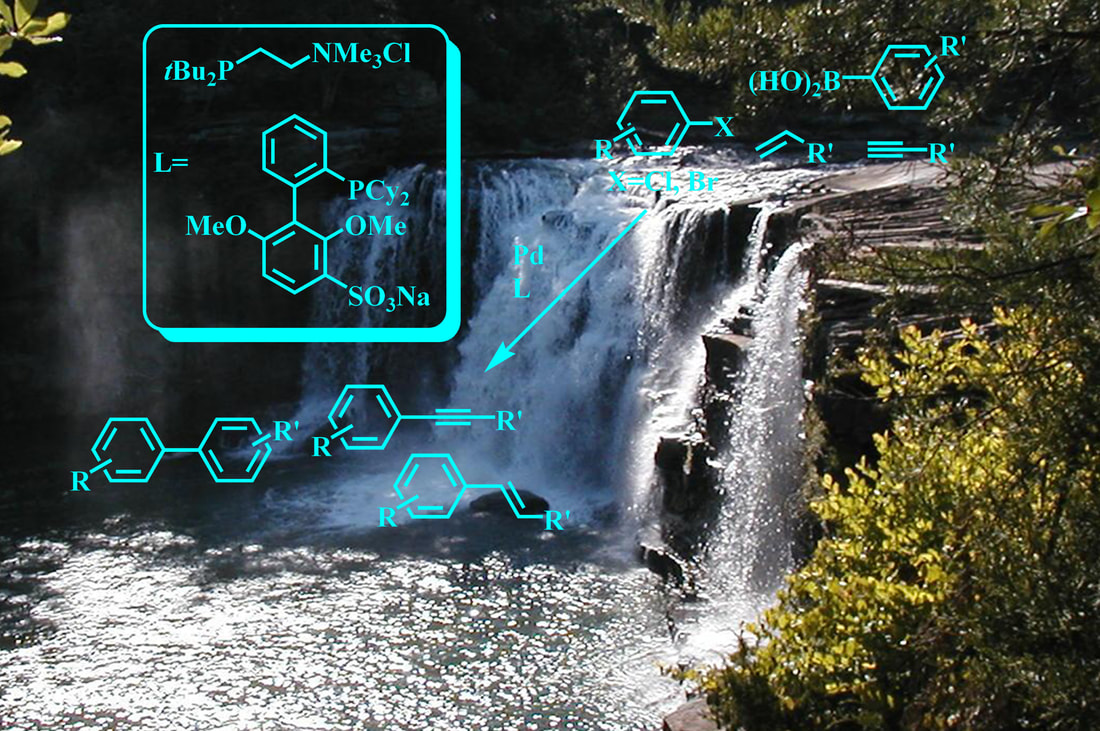
17. "Beyond TPPTS: New Approaches to the Development of Efficient Palladium-Catalyzed Aqueous-Phase Cross-Coupling Reactions," Kevin H. Shaughnessy, Eur. J. Org. Chem., 2006, 1827-1835. Cover Artwork.
• Invited review. •Top 10 most downloaded article in March and April 2006. Water is an attractive solvent for metal catalyzed reactions with both economic and environmental sustainability benefits. In addition, the use of a hydrophilic catalyst in an aqueous-biphasic solvent system provides the opportunity to easily recovery, and potentially recycle, the catalyst species. The feasibility of cross-coupling reactions catalyzed by hydrophilic palladium catalysts was first demonstrated using sulfonated analogs of triphenylphosphine, such as tri(3-sulfonatophenyl)phosphine trisodium salt (TPPTS). Catalysts derived from TPPTS and other structurally similar hydrophilic phosphines are generally effective only with aryl iodides and activated aryl bromides. This microreview will focus on more recently developed hydrophilic ligands with increased steric demand and electron donating abilities that are effective in coupling reactions of aryl bromides and chlorides under mild conditions in aqueous solvents. |
|
|
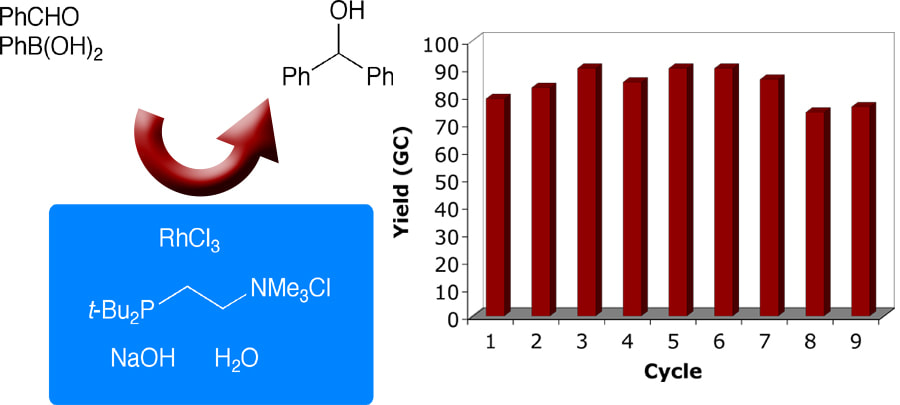
16. "t-Bu-Amphos-RhCl3•3H2O: A Highly Recyclable Catalyst System for the Cross-Coupling of Aldehydes and Aryl- and Vinylboronic Acids in Aqueous Solvents," Huang, R.; Shaughnessy, K. H., Chem. Commun., 2005, 4484-4486.
The combination of t-Bu-Amphos and RhCl3•H2O gave the first highly recyclable catalyst for the coupling of aryl- and vinylboronic acids with aldehydes in aqueous solvents. |
|
|
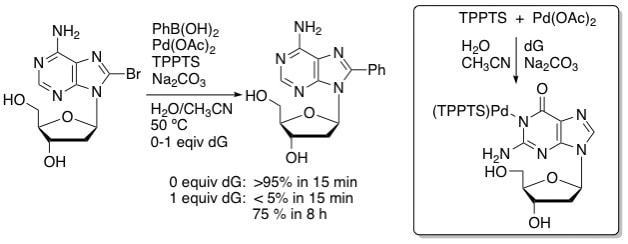
15. "Inhibitory Effects of the Guanine Moiety on Suzuki Couplings of Unprotected Halonucleosides in Aqueous Media," Western, E. C.; Shaughnessy, K. H., J. Org. Chem., 2005, 70, 3678-3688.
In the Suzuki arylations of unprotected halonucleosides in aqueous media, 8-bromo-2¢-deoxyguanosine (8BrdG) couplings were slower to reach completion than the corresponding 8-bromo-2¢-deoxyadenosine (8BrdA) couplings. The guanine moiety has an acidic proton, which under our Suzuki conditions (pH = 10), may be deprotonated to give an anion that can coordinate to palladium. The possibility that guanine coordination was responsible for the observed slower rates was explored using additive experiments, in which non-halogenated nucleosides were added to the Suzuki coupling reaction of 8BrdA or 4-bromotoluene and PhB(OH)2 and the reaction progress monitored by HPLC or GC. Adding dG slowed these reactions and an induction period was observed. The addition of dA or 1-methyl-2¢-deoxyguanosine (1MedG) to these couplings did not affect the rate of conversion to product. Guanine coordination was further explored using 13C and 31P NMR spectroscopy, which implies that guanine is coordinating to palladium through N-1 or O-6, or both. Furthermore, the presence of dG inhibited the formation of the active palladium(0) catalytic species, which may account for both the observed induction period and the sluggishness of reactions where guanine is involved. |
|
|
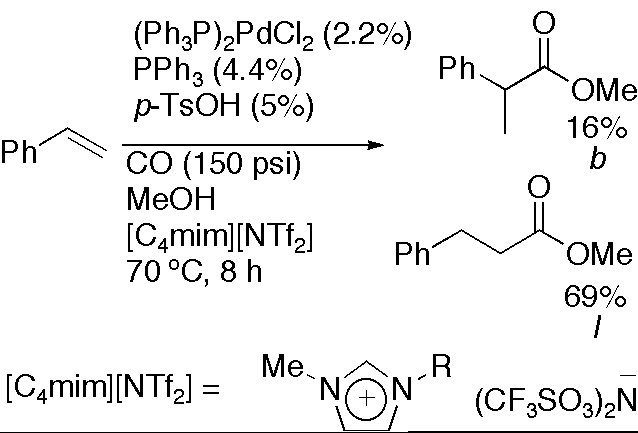
14. "Palladium-Catalyzed Hydroesterification of Styrene Derivatives in the Presence of Ionic Liquids," Klingshirn, M. A.; Rogers, R. D.; Shaughnessy, K. H. J. Organomet. Chem. 2005, 690, 3620-3626.
• Special Ionic Liquids Issue • #25 most downloaded article for 3rd quarter of 2005 The palladium-catalyzed hydroesterification of styrene in the presence of various ionic liquids has been successfully demonstrated with the presence of the ionic liquids providing high yields, high linear to branched ratios, and ease of catalyst recycling. |
|
|
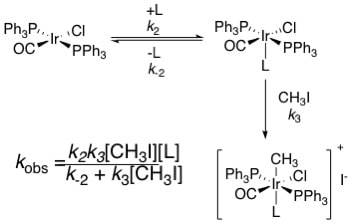
13. "Kinetic Study of the Oxidative Addition of Methyl Iodide to Vaska's Complex in Ionic Liquids," P'Pool, S. J.; Klingshirn, M. A.; Rogers, R. D.; and Shaughnessy, K. H. J. Organomet. Chem. 2005, 690, 3522-3528.
• Special issue on Ionic Liquids Oxidative addition of methyl iodide to Vaska's complex in the ionic liquids 1-butyl-3-methylimidazolium triflate [C4mim][OTf], [C4mim] bis(trifluormethylsulfonyl)imide [Tf2N], and N-hexylpyridinium [C6pyr][Tf2N] occurred cleanly to give the expected Ir(III) oxidative addition product. Pseudo-first order rate constants were determined for the oxidative addition reaction in each solvent ([Vaska's] = 0.25 mM, [CH3I] = 37.5 mM). The observed rate constants under these conditions were 5-10 times slower than the rate seen in DMF. At high methyl iodide concentrations (> 23 mM), the expected first order dependence on methyl iodide was not observed. In each ionic liquid, there was no change in the reaction rates within experimental error over the methyl iodide concentration range of 23 mM to 75 mM. At lower methyl iodide concentration, a decrease in rate was observed in [C4mim][Tf2N] with decreasing concentration of methyl iodide. |
|
|
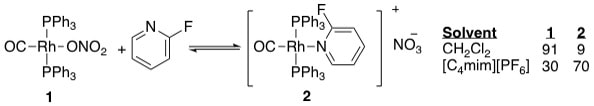
12. "Promoting Effect of Ionic Liquids on Ligand Substitution Reactions." Sliger, M. D.; Traylor, R. K.; McNeill, J., III; Young, S. H.; Hoffman, N. W.; Klingshirn, M. A.; Rogers, R. D.; and Shaughnessy, K. H. J. Organomet. Chem. 2005, 690, 3540-3545.
• Special issue on Ionic Liquids N-hexylpyridinium bistrifylimide ([C6pyr][Tf2N] and 1-butyl-3-methylimidazolium hexafluorophosphate ([C4mim][PF6]) promoted the displacement of anionic ligands by pyridine derivatives in trans-(Ph3P)2Rh(CO)X (X=NO3-, C6F5CO2-) to a much greater extent than methylene chloride. Thus, addition of a slight excess of 2-fluoropyridine to trans-(Ph3P)2Rh(CO)NO3 in [C4mim][PF6] gave an equilibrium ratio of 30:70 trans-(Ph3P)2Rh(CO)NO3:[trans-(Ph3P)2Rh(CO)(2-fluoropyridine)]NO3-, while the ratio was 91:9 in methylene chloride. 11. "Palladium-Catalyzed Cross-Coupling in Aqueous Media: Recent Progress and Current Applications." Shaughnessy, K. H.; DeVasher, R. B. Curr. Org. Chem. 2005, 9, 585-604. • Invited review for a special issue on modern transition-metal catalyzed reactions. Water has attracted significant attention as an alternative solvent for homogeneous metal-catalyzed reactions because it is a non-toxic, nonflammable solvent that can allow for simplified recovery of homogeneous catalysts. The use of water as a solvent in palladium-catalyzed cross-coupling reactions is of great academic and industrial interest. Palladium-catalyzed coupling reactions are powerful methods to make carbon-carbon and carbon-heteroatom bonds under mild conditions. Separation of the catalyst from the organic product can be very difficult, however. Constraining the catalyst to the aqueous-phase of an aqueous-biphasic solvent system can potentially simplify catalyst recovery. This review will focus on recent developments in the design and application of aqueous-phase, palladium-catalyzed coupling reactions over the period from 2000 through late 2004. A variety of new hydrophilic ligand architectures have been introduced recently that provide significantly more active catalysts towards industrially relevant aryl bromides and chlorides. |
|
|
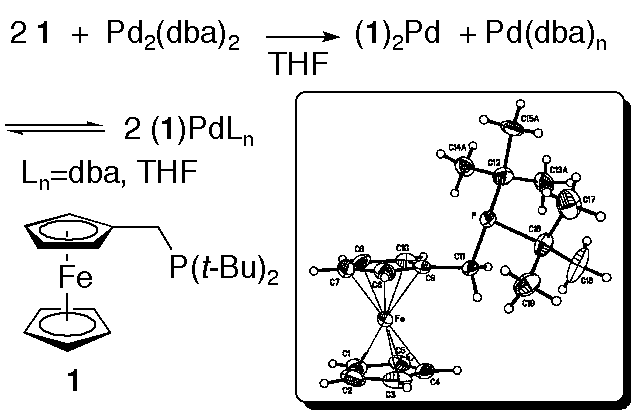
10. "Di-t-butyl(ferrocenylmethyl)phosphine: air-stability, structural characterization, coordination chemistry, and application to palladium-catalyzed cross-coupling reactions." Sliger, M. D.; Shaughnessy, K. H.; Broker, G. A.; Griffin, S. T.; Rogers, R. D. J. Organomet. Chem., 2005, 690, 1478-1486.
• #7 most downloaded article for the 1st quarter of 2005 • Top 25 download from JOMC for 2005 Di-t-butyl(ferrocenylmethyl)phosphine (1) has been isolated and structurally characterized. This ligand was found to be reasonably air stable as a solid and it has been shown to possess electron donating ability similar to that of tri-i-propylphosphine. A palladium catalyst bearing this ligand performed room temperature Suzuki-Miyaura coupling reactions with aryl bromides. Modest Heck coupling reactivity with aryl bromides was also observed at 100 ºC. Complexation of 1 with Pd2(dba)3 led to formation of (1)2Pd0. Addition of 4-bromoanisole to solutions containing both 1 and Pd2(dba)3 led to formation of an oxidative addition product when 1:Pd ratios were ≤ 1. With a 2:1 ratio of 1:Pd, monophosphine complex formation and oxidative addition were significantly inhibited. |
|
|
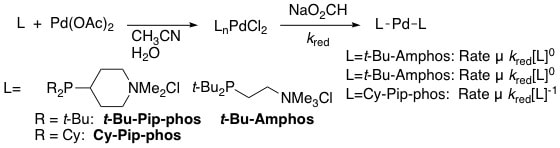
9. "Experimental and Computational Study of Steric and Electronic Effects on the Coordination of Sterically Demanding, Water-Soluble Alkylphosphines to Palladium under Reducing Conditions: Correlation to Catalytic Activity." DeVasher, R. B.; Spruell, J. M.; Dixon, D. A.; Broker, G. A.; Griffin, S. T.; Rogers, R. D.; Shaughnessy, K. H. Organometallics, 2005, 24, 962-971.
Sterically demanding, water-soluble alkylphosphine ligands 2-(di-t-butylphosphino)ethyltrimethylammonium chloride (t-Bu-Amphos) and 4-(di-t-butylphosphino)-N,N-dimethylpiperidinium chloride (t-Bu-Pip-phos) in combination with palladium salts provided active catalysts for the cross-coupling of aryl halides under mild conditions in aqueous solvents, whereas 4-(dicyclohexylphosphino)-N,N-dimethylpiperidinium chloride (Cy-Pip-phos) gave a less active catalyst. Catalyst activity increased with increasing cone angle of the ligands, but the c electronic parameter determined from the symmetric C-O stretching frequency of LNi(CO)3 did not correlate with catalyst activity. Catalyst activity correlated with other calculated electronic parameters, such as the HOMO-LUMO energy gap of the ligand and the HOMO energy level of the LPd(0) species. Multinuclear NMR spectroscopic studies showed that t-Bu-Amphos and t-Bu-Pip-phos rapidly form L2Pd(0) (L = t-Bu-Amphos or t-Bu-Pip-phos) complexes when reacted with Pd(OAc)2 under reducing conditions over a range of L:Pd ratios. In contrast, the coordination chemistry of Cy-Pip-phos depended on the Cy-Pip-phos:Pd ratio. At a ≤ 1:1 Cy-Pip-phos:Pd ratio, rapid formation of L2Pd(0) occurred. At higher L:Pd ratios, initial formation of trans-(Cy-Pip-phos)2PdCl2 was observed followed by slow reduction to the Pd(0) complex. |
|
|
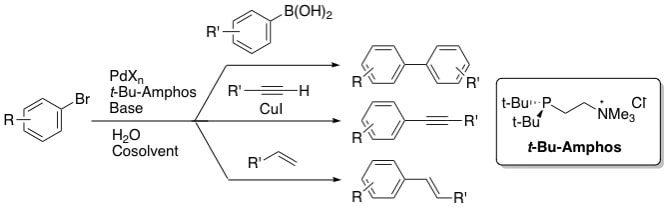
8. "Aqueous-Phase, Palladium-Catalyzed Cross-Couplings of Aryl Bromides Under Mild Conditions Using Water-Soluble, Sterically Demanding Alkylphosphines." DeVasher, R. B.; Moore, L. R.; Shaughnessy, K. H. J. Org. Chem. 2004, 69, 7919-7927.
Sterically demanding, water-soluble alkylphosphines have been used in combination with various palladium salts in Suzuki, Sonogashira, and Heck couplings of aryl bromides under mild conditions in aqueous solvents. The t-butyl-substituted ligands 2-(di-t-butylphosphino)ethyltrimethylammonium chloride (t-Bu-Amphos) and 4-(di-t-butylphosphino)-N,N-dimethylpiperidinium chloride (t-Bu-Pip-phos) in combination with palladium(II) salts were found to give catalysts that were significantly more active than catalysts derived from tri-(3-sulfonatophenyl)phosphine trisodium (TPPTS). Suzuki couplings of unactivated aryl bromides occurred efficiently at room temperature in water/acetonitrile and water/toluene biphasic mixtures or in neat water. Notably, Suzuki couplings of hydrophilic aryl bromides gave high yields without using organic solvents for the reaction or purification. This methodology has been applied to a highly efficient synthesis of diflunisal. The catalyst derived from t-Bu-Amphos was recycled three times in Suzuki couplings in water/toluene before catalyst activity began to significantly drop. The average yield of four cycles was > 80%/cycle. Heck and Sonogashira couplings were carried out under mild conditions (50 ˚C and 80 ˚C respectively) with unactivated aryl bromides to give coupled products in high yield. |
|
|
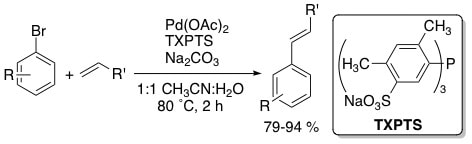
7. "Efficient aqueous-phase Heck and Suzuki couplings of aryl bromides using tri(4,6-dimethyl-3-sulfonatophenyl)phosphine trisodium salt (TXPTS)." Moore, L. R. and Shaughnessy, K. H. Org. Lett. 2004, 6, 225-228.
Sterically demanding, sulfonated arylphosphines TXPTS and TMAPTS have been applied to the aqueous-phase Heck and Suzuki coupling of aryl bromides. TXPTS provides good yields of Heck coupling products from aryl bromides at 80 ˚C, while both TMAPTS and TPPTS gave significantly less active catalysts. TXPTS is the first ligand to promote the aqueous-phase Heck coupling under such mild conditions. Both TXPTS and TMAPTS provide active catalysts for Suzuki couplings of aryl bromides at 50 ˚C. |
|
|
6. “Synthesis, Properties and NMR Studies of a C8-Phenylguanine Modified Oligonucleotide that Preferentially Adopts the Z-DNA Conformation.” Gannett, P. M.; Heavner, S.; Daft, J. R. Shaughnessy, K. H.; Epperson, J. D.; Greenbaum, N. L. Chem. Res. Tox. 2003, 16, 1384-1395.
Carcinogenic aryl hydrazines produce C8-arylated purine adducts. The effect of these adducts on DNA conformation and their role in hydrazine carcinogenesis are unknown. Here, we describe a new synthetic route to produce these adducts that is also compatible with the synthesis of the corresponding phosphoramidites needed for oligonucleotide synthesis. Two oligonucleotides were prepared, an unmodified oligonucleotide, d(5¢CGCGCGCGCG3¢), and a C8-phenylguanine modified oligonucleotide, d(5¢CGCGCG*CGCG3¢) (G* ) 8-phenylguanine). These oligonucleotides were compared using thermal denaturation, circular dichroism, NMR, and molecular modeling. The phenyl modification destabilizes the B DNA form and stabilizes the Z DNA form such that the B:Z ratio is near one under physiological conditions. In light of recent studies that show a role for Z DNA in gene expression and cell transformation, Z DNA stabilization by C8-arylguanine formation from aryl hydrazines may be relevant to their role in carcinogenesis. |
|
|
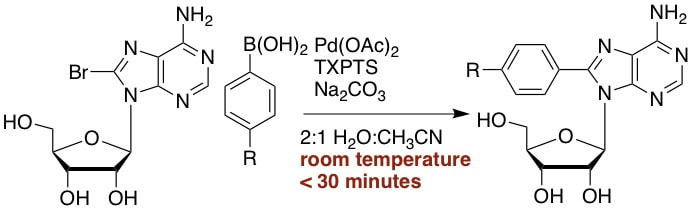
5. “Efficient One-Step Suzuki Arylation of Unprotected Halonucleosides using Water-Soluble Palladium Catalysts,” Western, E. C.; Daft, J. R.; Johnson, E. M., II; Gannett, P. M.; Shaughnessy, K. H. J. Org. Chem., 2003, 68, 6767-6774.
Modification of nucleosides to give pharmaceutically active compounds, mutagenesis models, and oligonucleotide structural probes continues to be of great interest. The aqueous-phase modification of unprotected halonucleosides is reported herein. Using a catalyst derived from tris(3-sulfonatophenyl)phosphine (TPPTS) and palladium acetate, 8-bromo-2'-deoxyguanosine (8-BrdG) is coupled with arylboronic acids to give 8-aryl-2'-deoxyguanosine adducts (8-ArdG) in excellent yield in a 2:1 water:acetonitrile solvent mixture. The TPPTS ligand was found to be superior to water-soluble alkylphosphines for this coupling reaction. The coupling chemistry has been extended to 8-bromo-2'-deoxyadenosine (8-BrdA) and 5-iodo-2'-deoxyuridine (5-IdU), as well as the ribonucleosides 8-bromoguanosine and 8-bromoadenosine. Good to excellent yields of arylated adducts are obtained in all cases. Using tri-(4,6-dimethyl-3-sulfonatophenyl)phosphine (TXPTS), the Suzuki coupling of 8-BrdA and 5-IdU can be accomplished in less than one hour at room temperature. This methodology represents an efficient and general method for halonucleoside arylation that does not require protection of the nucleoside prior to arylation. |
|
|
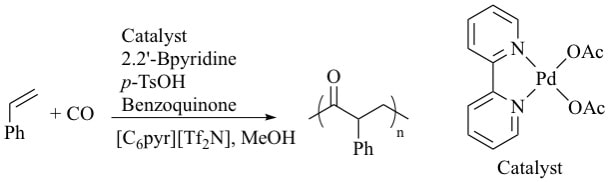
4. "Polar, Non-Coordinating Ionic Liquids as Solvents for Coordination Polymerization of Olefins," Shaughnessy, K.H.; Klingshirn, M. A.; P'Pool, S. J.; Holbrey, J. D.; Rogers, R. D. ACS Symposium Series 856: Ionic Liquid as Green Solvents: Progress and Prospects, Rogers, R. D.; Seddon, K. R. (Eds), American Chemical Society, Washington, DC, 2003, 300-313
Increased polymerization yield is observed in the copolymerization of styrene and CO in ionic liquids (ILs) compared to commonly used molecular solvents using palladium catalysts. Conditions for the copolymerizations were optimized and the effect of changes in the cation and anion of the IL solvent were determined. These results suggest that polar, non-coordinating ILs accelerate olefin polymerization catalyzed by electrophilic, charge-separated catalyst species. |
|
|
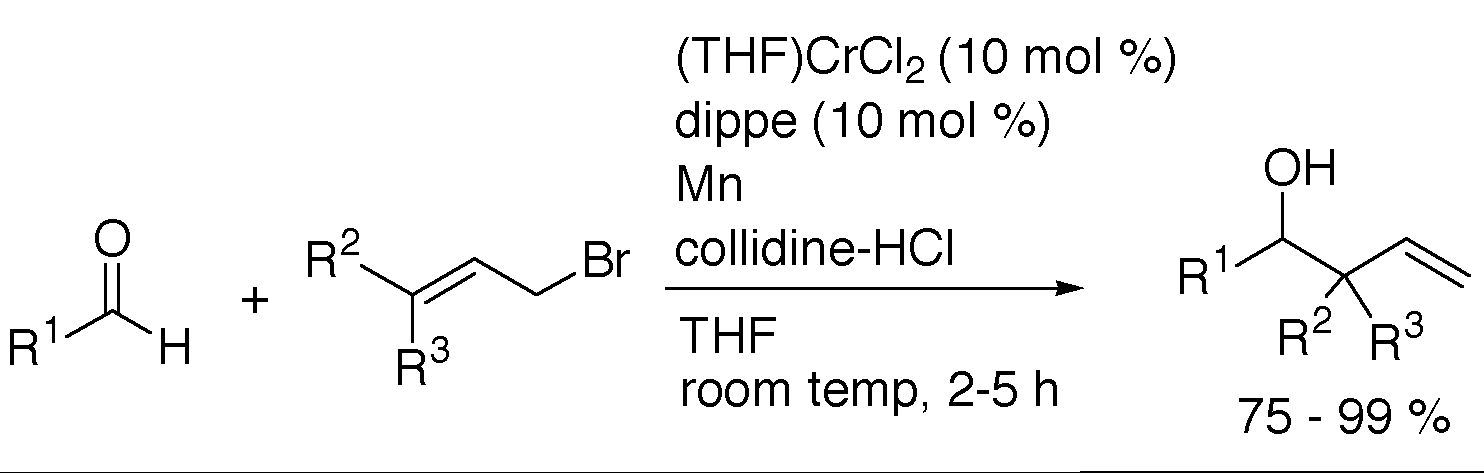
3. "Acid-Mediated, Chromium-Catalyzed Allylation of Aldehydes," K. H. Shaughnessy and R. Huang, Synth. Comm., 2002, 32, 1923-1928.
Allyl bromides are efficiently coupled with aryl and aliphatic aldehydes in the presence of manganese metal, collidine hydrochloride, bis(diisopropylphosphino)ethane and chromium dichloride. Homoallylic alcohols are isolated in good to excellent yields directly from the reaction mixture. |
|
|
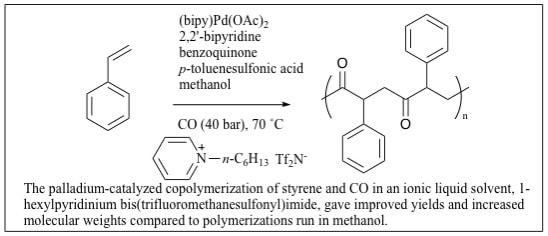
2. "Polar, Non-Coordinating Ionic Liquids as Solvents for the Alternating Copolymerization of Styrene and CO Catalyzed by Cationic Palladium Catalysts." Klingshirn,M. A.; Broker G. A.; Holbrey, J. D.; Rogers, R. D.; and Shaughnessy, K. H. Chem. Commun. 2002, 1394-1395.
The palladium-catalyzed copolymerization of styrene and CO in an ionic liquid solvent, 1-hexylpyridinium bis(trifluoromethanesulfonyl)imide, gave improved yields and increased molecular weights compared to polymerizations run in methanol. |
|
|
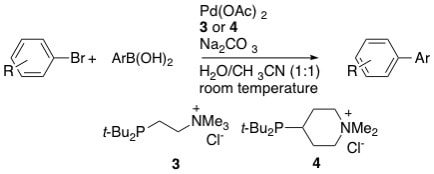
1. "Sterically Demanding, Water-Soluble Alkylphosphines as Ligands for High Activity Suzuki Couplings in Aqueous Solvents" Shaughnessy, K. H. and Booth, R. S. Org. Lett., 2001, 3 2757-2759.
Sterically demanding, water-soluble alkylphosphines have been found to give highly active catalysts for Suzuki coupling of aryl bromides in aqueous solvents. A variety of aryl bromides and boronic acids were coupled in excellent yield. Turnover numbers up to 734 000 mmol/mmol Pd have been achieved under mild conditions. |
|
Post-Doctoral Publications
- "Screening of Homogeneous Catalysts by Fluorescence Resonance Energy Transfer. Identification of Catalysts for Room Temperature Heck Reactions," J. P. Stambuli, S. R. Stauffer, K. H. Shaughnessy, J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 2677-2678.
- "Room Temperature Palladium-Catalyzed Amination of Aryl Bromides and Chlorides and Extended Scope of Aromatic C-N Bond Formation with a Commercial Ligand." J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy, L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575-5580.
- "A Fluorescence Assay for High-Throughput Screening of Coupling Reactions. Application to Heck Coupling Processes." K. H. Shaughnessy, P. Kim, J. F. Hartwig, J. Am. Chem. Soc. 1999, 121, 2123-2131.
- "Palladium Catalyzed Inter-and Intramolecular a-Arylation of Amides. Application of Intramolecular Amide Arylation to the Synthesis of Oxindoles." K. H. Shaughnessy, B. C. Hamann, J. F. Hartwig, J. Org. Chem., 1998, 63, 6546-6553.
- "Transition metal-catalyzed process or preparing N-aryl compounds," J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy, L. M. Alcazar-Roman (Yale University), US Patent 6,100,398 (2000)
Graduate Publications
- "Enantio- and Diastereoselective Catalytic Carboalumination of 1-Alkenes and α,ω-Dienes with Cationic Zirconocenes: Scope and Mechanism." K. H. Shaughnessy and R. M. Waymouth, Organometallics, 1998, 17, 5728-5745.
- "Highly Regioselective Cyclocarboxylation of Nonconjugated Dienes Catalyzed by Palladium." K. H. Shaughnessy and R. M. Waymouth,Organometallics, 1997, 16, 1001-1007.
- "Carbometalation of α,ω-Dienes and Olefins Catalyzed by Zirconocenes." K. H. Shaughnessy and R. M. Waymouth, J. Am. Chem. Soc. 1995, 117, 5873-5874.
Undergraduate Publcations
- "A Facile Method for the Preparation of Functionalized 2-Halo-1-olefins." Zhu, L.; Shaughnessy, K. H.; Rieke, R. D. Synth. Comm., 1993, 23, 525-529.
Patents at UA
- "Synthesis of a Hierarchically Porous Monoliths By A Cogelation Method," Bakker, M. G.; Kotbagi, T. V.; Shaughnessy, K. H. US Patent, 10,195,587, February 5th, 2019.
- "Catalysis by Metal Nanoparticles Dispersed Within a Hierarchically Porous Carbon" Material, Bakker, M.G.; Sayler, F.M.; Shaughnessy, K.H., US Patent 9,233,366, January 12, 2016.
Books and Chapters at UA
- Hartwig, J. F.; Shaughnessy, K. H.; Shekhar, S.; Green, R. A. "Palladium-Catalyzed Amination of Aryl Halides," in Organic Reactions, vol 100, Denmark, S. ed., John Wiley and Sons, 2019, vol. 100, pp 853-958.
- Shaughnessy, K. H. "Introduction of water-solubility in palladacycles and their catalytic application," in Palladacycles: Catalysis and Beyond, Kapdi, A.; Maiti, D. eds., Burlington: Elsevier, 2019, 225-247, ISBN 978-0-12-815505-9
- Shaughnessy, K. H. "Application of water soluble palladium-catalyst systems for introduction of C-C bonds in nucleosides," in Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides, Kapdi, A. R.; Maiti, D.; Sanghvi, Y. S., eds. Elsevier, 2018, pp 247-268.
- Shaughnessy, K. H.; Ciganek, B.; DeVasher, R. B. Copper-Catalyzed Amination of Aryl and Alkenyl Electrophiles, John Wiley & Sons, Hoboken, NJ, 2017, 696 pp.
- Shaughnessy, K. H.; Ciganek, B.; DeVasher, R. B. " Copper-Catalyzed Amination of Aryl and Alkenyl Electrophiles," in Organic Reactions, vol. 85, Denmark, S. Ed., John Wiley and Sons, 2014, 1-668.
- Shaughnessy, K. H. "Greener Approaches Cross-Coupling," in New Trends in Cross-Coupling: Theory and Applications. Colacot, T. J. ed., RSC Publishing, 2015, pp 645-696.
- Shaughnessy, K. H. "Cross-Coupling Reactions in Aqueous Media," in Palladium-Catalyzed Cross-Coupling Reactions - Practical Aspects and Future Developments, Molnár, Á. Ed., Wiley-VCH, 2013, pp 235-286. ISBN: 978-3-527-33254-0
- Shaughnessy, K. H. "Metal-Catalyzed Cross-Couplings of Aryl Halides to Form C–C Bonds in Aqueous Media," in Metal-Catalyzed Reactions in Water, Diexnuf, P.; Cadierno, V. Eds., Wiley-VCH, 2013, pp 1-46. ISBN: 978-3-527-33188-8
Published Abstracts and Non-Peer-Reviewed Publications at UA
- "Tri(neopentyl)phosphine" Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis (e-EROS), 2014.
- "1-Trimethylammonium-2-(di-t-butyl)phosphino)ethane chloride, t-Bu-Amphos," Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis (e-EROS), published online April 22, 2013
- Sayler, F. M.; Grano, A. J.; Scogin, W.; Sanders, P.; Smått, J.-H.; Shaughnessy, K. H.; Bakker, M. G. "Formation and Applications of Hierarchically Porous Carbon, Metals and Metal Oxides Formed by Nanocasting", MRS Symp. Proc., 2012, 1389, 1-8.
- "Bis(1,1-dimethylethyl)2,2-dimethylpropylphosphine," Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis (e-EROS), published online October 15, 2010
- "2-(Pentafluorophenyl)-1,3-bis(2,4,6-trimethylphenyl)-imidazolidine," Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis(eEros), 2008, published online September 15th, 2008.
- "Tri-tert-butylphosphonium tetrafluoroborate," Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis (eEros), 2008, published online March 14th, 2008.
- “Palladium, bis(tris-(1,1-dimethylethyl)phosphine,” Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis (eEros), 2008, published online March 14th, 2008.
- "On Comprehensive Organic Reactions in Aqueous Media by Chao-Jun Li and Tak-Hang Chan" Shaughnessy, K. H. " Angew. Chem. Int. Ed. 2008,47, 1988. (Book Review)
- "Tri-tert-butylphosphine," Shaughnessy, K. H. Electronic Encyclopedia of Reagents for Organic Synthesis (eEros), 2007, published online March 15th, 2007.
- "Coordination Polymerization of Alkenes in Ionic Liquids." Shaughnessy, K. H.; P'Pool, S. J.; Klingshirn, M. A.; Rogers, R. D. Polymer Prepr. 2004, 45, 317-318.
- "Transition Metal Catalyzed CO/Olefin Co-Polymerization in Room Temperature Ionic Liquids," Holbrey, J. D.; Shaughnessy, K. H.; Klinshirn, M. A.; Broker, G. A.; Rogers, R. D. in Molten Salts XIII, Delong, H. C.; Bradshaw, R. W.; Matsunaga, M.; Stafford, G. R.; Trulove, P. C. Eds. 2002, 2002-19, pp 213-223, The Electrochemical Society Proceedings Series, Pennington, NJ.
Student Theses and Dissertations
PhD Dissertations
2022
MS Theses
2019
Senior Theses
2010
2022
- Corrie J. Burlas. An Evaluation of the Ligand Effects on Neopentyl Phosphine Derived Precatalysts for N-Arylation Reactions and for the Cross-Coupling of Aryl Acetylenes with Propargyl Alcohols and Amides
- Benjamin R. Headford. Development of Modular Synthetic Pathways Towards Novel P‑Unsymmetric Bidentate Ligands
- Jordan K. Pierce. Synthesis and Application of Air-Stable Secondary Phosphine Oxide, Oxime, and Amine-Phosphine Palladium(II) Complexes in Transfer Hydrogenation and Cross-Coupling Reactions
- Kerry L. Barnett. Air-Stable Palladium(II) Precatalysts: Synthesis, Properties, and Applications in Cross-Coupling Reactions
- Huaiyuan "Ethan" Hu. Part A: Arenepolythiols Synthesis And Doping Study Of Semiconducting Metal-Organic Framework; Part B: Mechanistic Study Of The Bis(Trineopentylphosphine)Palladium Catalyzed Buchwald-Hartwig Amination
- Jane N. Moore. Synthesis and Application of Sterically Flexible and Water-Soluble Phosphine Ligands in Palladium Catalysis
- William Scott Brown, Design and Synthesis of Phosphine Ligands for Palladium-Catalyzed Coupling Reactions
- Lensey L. Hill, Investigations of Cross Coupling Reactions: Synthesis and Scope of New Neopentyl Phosphine Ligands and Pre-formed Palladium Catalysts
- Joon Hyung Cho, Part I. Palladium -Catalyzed Alkenylation and Alkynylation of Nucleosides in Aqueous Media. Part II. Palladium-Catalyzed Carbon-Selenium Bond formation for Unsymmetrical Diselenides
- Lucas R. Moore, Ligand Design and Application Toward Palladium-Catalyzed Cross-Coupling Reactions
- S. Justin P'Pool, Polar, Weakly Coordinating Ionic Liquids as Solvents for Fundamental Organometallic Reactions
- Rongcai Huang, Part I. Asymmetric Hydrosilylation of Acetophenone Using Titanium Catalysts; Part II. Synthesis of Water-Soluble Palladacycles and Their Applications to the Suzuki Reaction; Part III. Rhodium-Catalyzed Addition of Aryl and Alkenylboronic Acids to Aldehydes
- Elizabeth C. Western, Palladium-Catalyzed Modification of Halonucleosides: Methodology Development and Mechanism Determination
•Received University of Alabama Outstanding Dissertation Award
- Rebecca B. DeVasher, Water-Soluble Alkylphosphines as Ligands in Palladium-Ctalyzed Aqueous-Phase Cross-Coupling Reactions: Mechanistic Advances and Catalytic Activity
MS Theses
2019
- Ashley M. Gilliam, Using Ketones as Catalyst Activators in Palladium-Catalyzed Cross-Coupling Reactions
- Elizabeth C. Western, Efficient Modification of Halonucleosides Using Suzuki-Miyaura Coupling in Aqueous Media
•Named Outstanding Thesis by College of Arts and Sciences
Senior Theses
2010
- Hannah K. Box, The Synthesis of Water-Soluble Phosphines and their Application to Recyclable, Aqueous Phase Palladium Catalysts
